Информация о COVID-19

На портале представлена современная биомедицинская информация о механизмах развития патологического процесса при COVID-19, влиянии этой инфекции на биологические процессы в организме, о фармакологических мишенях терапевтического воздействия, об антикоронавирусных препаратах, применяемых для лечения заболевания, а также об исследованиях с целью репозиционирования лекарств для терапии COVID-19.
Биологические процессы
Биологические процессы в организме человека на которые оказывает влияние SARS-CoV-2
Мы прилагаем все усилия по актуализации представленных данных. (Выберите интересующую вкладку ниже для получения более подробной информации)
Основные мишени препаратов прямого противовирусного действия для терапии инфекции SARS-CoV-2
В геноме SARS-CoV-2, содержащем около 30000 нуклеотидов, определено 17 открытых рамок считывания (табл. 1) [1-3], которые кодируют до 30 белков [4, 5]. Белки SARS-CoV-2, а также взаимодействующие с ними белки человека рассматриваются как фармакологические мишени, воздействие на которые может быть использовано для терапии новой коронавирусной инфекции.
| Рамка считывания | Начальная позиция в РНК | Конечная позиция в РНК | Длина аминокислотной последовательности | UniProt AC |
|---|---|---|---|---|
| ORF1a | 266 | 13483 | 4405 | P0DTC1 |
| ORF1ab | 266 | 21555 | 7096 | P0DTD1 |
| S | 21563 | 25384 | 1273 | P0DTC2 |
| ORF3a | 25393 | 26220 | 275 | P0DTC3 |
| ORF3c | 25457 | 25582 | 41 | P0DTG1 |
| ORF3d | 25524 | 25697 | 57 | P0DTG0 |
| ORF3b | 25814 | 25882 | 22 | P0DTF1 |
| E | 26245 | 26472 | 75 | P0DTC4 |
| M | 26523 | 27191 | 222 | P0DTC5 |
| ORF6 | 27202 | 27387 | 61 | P0DTC6 |
| ORF7a | 27394 | 27759 | 121 | P0DTC7 |
| ORF7b | 27756 | 27887 | 43 | P0DTD8 |
| ORF8 | 27894 | 28259 | 121 | P0DTC8 |
| N | 28274 | 29533 | 419 | P0DTC9 |
| ORF9b | 28284 | 28577 | 97 | P0DTD2 |
| ORF9c | 28734 | 28955 | 73 | P0DTD3 |
| ORF10 | 29558 | 29674 | 38 | A0A663DJA2 |
| Рамка считывания | Начальная позиция в РНК | Конечная позиция в РНК | Длина аминокислотной последовательности | UniProt AC |
Примечание. Сформировано на основе записи NC_045512.2 в БД GenBank и публикации [5]. Данные по участкам, кодирующим структурные и вспомогательные белки, показаны жирным шрифтом и курсивом, соответственно. Координаты рамок включают терминирующие кодоны.
Различают три группы коронавирусных белков. Неструктурные (non-structural, nsp) белки образуются путем протеолитического процессинга полипротеина-предшественника, который кодируется рамкой ORF1a или ORF1ab. Эти белки участвуют в жизненном цикле вируса, обеспечивая, прежде всего воспроизводство вирусных РНК [6]. Структурные белки кодируются рамками S, E, M и N и формируют вирусные частицы (вирионы) на более поздних стадиях вирусного цикла [7, 8]. Белки S, E, M встроены в липидную оболочку вируса, а частицы N-белка связаны с РНК-геномом, образуя нуклеокапсид (Рисунок 1).
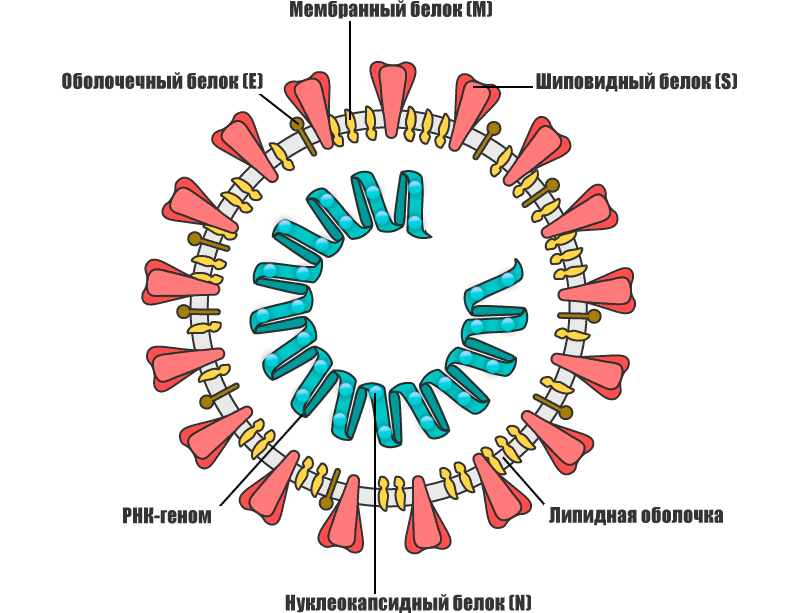
Ряд рамок кодирует вспомогательные белки (accessory proteins), которые не участвуют в воспроизводстве вируса, но взаимодействуют с хозяйскими белками, обеспечивая, в том числе, защиту от врожденного иммунитета [9, 10].
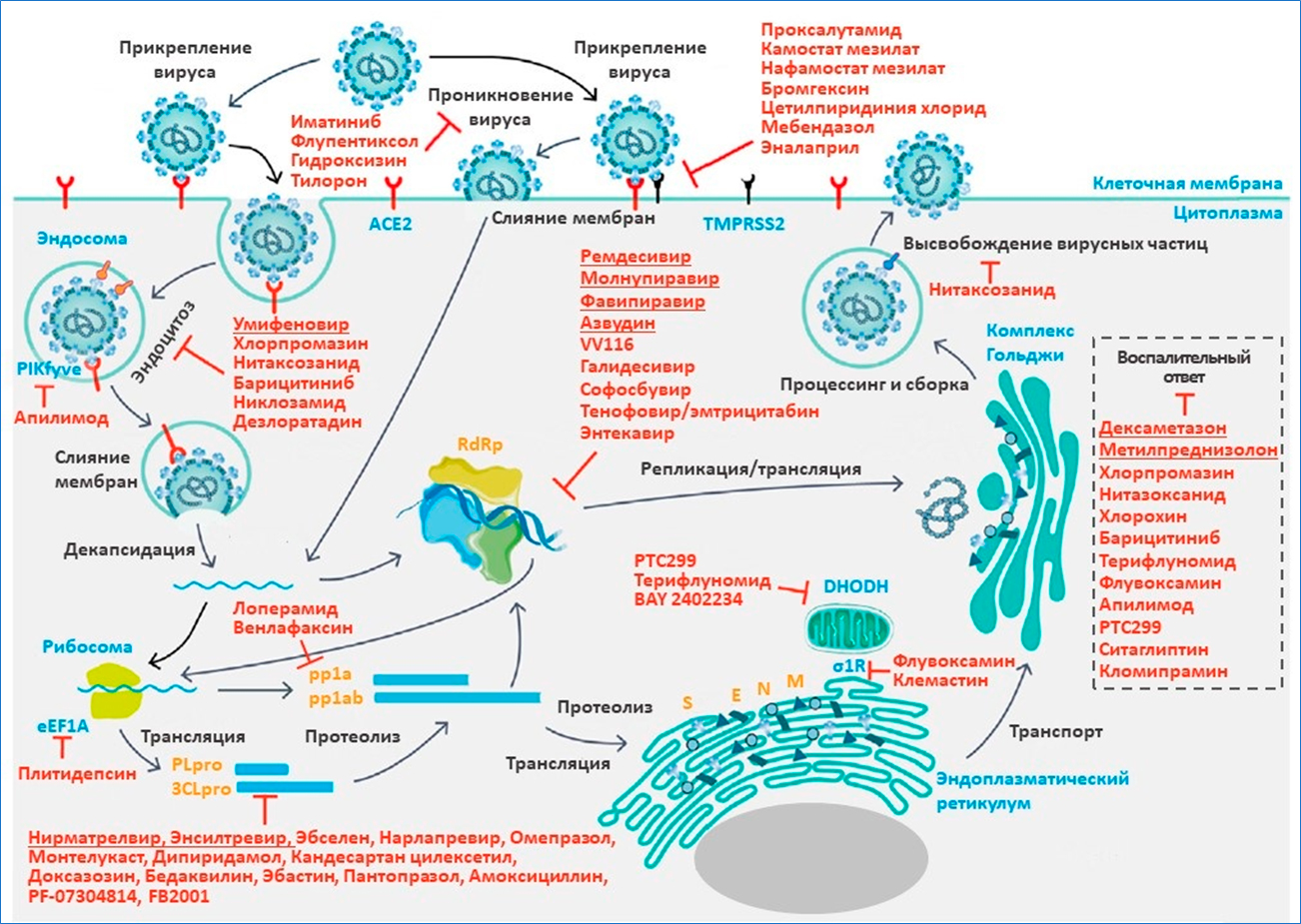
На рисунке 2 представлен жизненный цикл вируса SARS-CoV-2 с информацией о возможных механизмах действия некоторых рассматриваемых в литературе препаратов, потенциально обладающих антикоронавирусным действием.
Инфекционный цикл вируса SARS-CoV-2 состоит из нескольких этапов: прикрепление к клетке, проникновение в клетку, синтез РНК и белка, сборка вириона и высвобождение вирусных частиц из клетки.
1. Проникновение вируса в клетку
Прикрепление вириона с последующим проникновением вирусного материала осуществляется за счет взаимодействия вирусного гликопротеина S-белка с поверхностными структурами клетки. Он заякорен в вирусной оболочке в виде тримера. Каждый мономер состоит из двух субъединиц S1 (672 аминокислотных остатка) и S2 (588 остатков), которые образуются путем протеолитического расщепления из белка- предшественника S [12, 13]. Рецептор-связывающий домен S1 присоединяется к ангиотензинпревращающему ферменту 2-го типа (ACE2) человека [12]. Возможными рецепторами вируса также являются нейропилин [14], басигин [15], лектины [16], молекула повреждения почек 1 (KIM-1) [17] и ряд других белков [18, 19].
Множественность патологических процессов, происходящих в организме больного COVID-19, указывает на то, что число белков-рецепторов может быть гораздо больше, и список идентифицированных белков будет пополняться. Вместе с тем, опубликованные данные нуждаются в уточнении; так, например, в более поздней публикации роль басигина как возможного рецептора SARS-CoV-2 не была подтверждена [20].
Субъединица S1, индуцирующая выработку вируснейтрализующих антител, характеризуется высокой частотой мутаций, что создает проблемы при разработке средств вакцинопрофилактики и применении терапевтических антител. Это является важным обстоятельством, побуждающим к разработке ингибиторов взаимодействия S1 и ACE2 на основе фрагментов ACE2 [21; 22]. Осуществляется также поиск лигандов рецептор-связывающего домена на основе компьютерного моделирования с последующим тестированием in vitro [23].
Субъединица S2 обеспечивает слияние вирусной и клеточной мембран. Расщепление S2 одной из протеаз хозяина, таких как TMPRSS2 или катепсин L, освобождает пептид слияния в N-концевой части S2. Насыщенный гидрофобными остатками пептид слияния взаимодействует с клеточной мембраной, запуская структурные перестройки белка, приводящие к слиянию мембран и поступлению вирусного материала в цитоплазму либо через внешнюю мембрану клетки, либо путем клатрин-опосредованного эндоцитоза [12].
Субъединица S2 рассматривается как перспективная мишень, поскольку достаточно консервативна у разных вариантов вируса. Исследован ряд препаратов, которые действуют на подструктуры S2 и ингибируют слияние вирусной и клеточной мембран [24].
S-белок участвует в формировании многоядерных синцитиев [25], что способствует распространению вируса между клетками и, вероятно, позволяет ему уклоняться от иммунного ответа. Образование синцитиев зафиксировано у пациентов с тяжелой формой COVID-19.
Взаимодействие S-белка с ACE2 при проникновении вируса путем эндоцитоза обуславливает снижение экспрессии ACE2 в клетках легких за счет лизосомального расщепления. Это рассматривается как важный фактор патогенеза, связанный с нарушением регуляции ангиотензин-рениновой системы [26]. Выраженность указанного эффекта зависит от структурных особенной S-белка у конкретного вирусного штамма.
2. Формирование неструктурных белков и их функции
Слияние вирусной и клеточной мембран обеспечивает выход вирусного материала в цитоплазму. Освободившаяся геномная РНК связывается с рибосомами и используется непосредственно как матрица для трансляции с двух рамок считывания ORF1a и ORF1ab, занимающих около двух третей генома. Остальные рамки считывания служат шаблонами для синтеза субгеномных мРНК, которые, в свою очередь, выступают как матрицы для ситнтеза структурных и вспомогательных белков [27].
Рамка ORF1a совпадает с начальной частью ORF1ab. Использование механизма “программируемого сдвига рамки” (frameshifting) обеспечивает синтез двух вариантов белкового продукта (полипротеина-предшественника) pp1a и pp1аb длиной 4405 и 7096 аминокислотных остатков, соответственно [28]. Полипротеин аутокаталитически расщепляется вирусными протеазами PLpro и 3CLpro (рисунок 3, табл). Образовавшиеся неструктурные белки (non-structural proteins) обозначаются по порядку их расположения – Nsp1, …, Nsp16 [6]. Короткая последовательность Nsp11 в составе pp1a (4393-4405) вследствие сдвига рамки исчезает как отдельный участок в составе pp1ab, совпадая по первым девяти остаткам с N-концевой частью Nsp12.
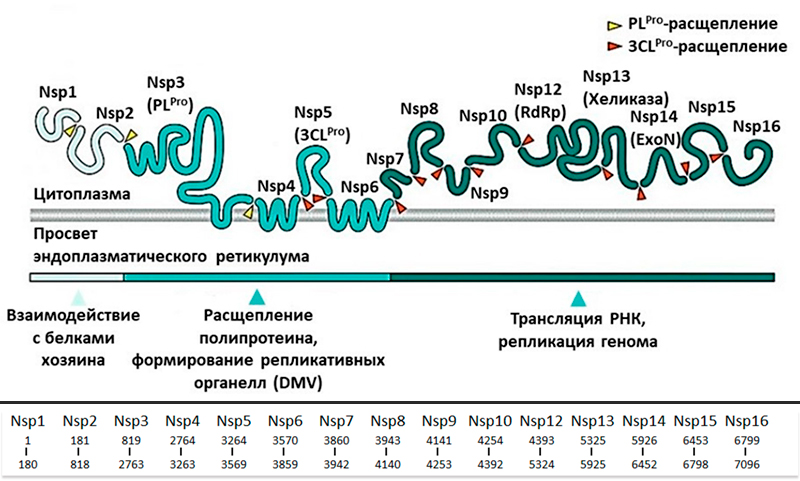
Белки Nsp1 и Nsp2 не включены непосредственно в репликацию вируса. Они взаимодействуют с клеточными структурами, отвечающими за синтез белка, что приводит, в том числе, к угнетению врожденного иммунитета.
Связываясь с субъединицей рибосомы 40S и вирусной мРНК, С-концевой участок Nsp1 перекрывает вход для хозяйских мРНК. В то же время, взаимодействие N-концевого домена nsp1 с 5’-нетранслируемой областью мРНК коронавируса открывает доступ для вирусных мРНК. Так происходит переключение белкового синтеза в сторону вирусных белков [29]. Помимо этого, Nsp1 участвует в расщеплении хозяйской мРНК [30], взаимодействует с белками, которые отвечают за экспорт клеточной мРНК из ядра [31]. Связывание Nsp1 c хозяйской праймазой [32] может нарушать репликацию ДНК человека.
Белок Nsp2 SARS-CoV-2 охарактеризован достаточно слабо. Делеция участка, кодирующего Nsp2 из геномов таких коронавирусов как SARS-CoV и MHV (вирус мышиного гепатита) приводит лишь к умеренному снижению вирусного титра [33]. Белки-ортологи Nsp2, экспрессируемые у SARS-CoV, SARS-CoV-2 и MERS, взаимодействуют с хозяйскими белками, вовлекаясь в такие процессы как регуляция трансляции, эндосомальный транспорт, биогенез рибосом [34]. Полагают, что Nsp2 участвует в угнетении трансляции хозяйских генов с участием микроРНК [35].
Nsp3 – самый большой неструктурный белок у всех коронавирусов [36]. У SARS-CoV-2 он содержит 1945 остатков. В Nsp3 выделяют 14 структурно-функциональных участков (Рисунок 4). Среди его многочисленных функций отмечают протеолитический процессинг полипротеина-предшественника, который производится доменом PLpro. Не менее важно и участие Nsp3 в формировании репликативных органелл.
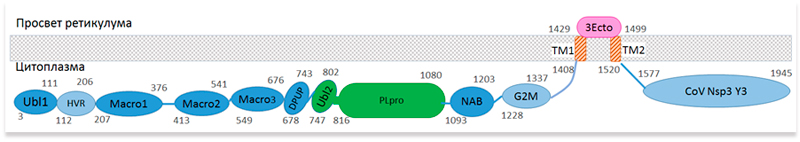
Домен PLpro – папаиноподобная цистеиновая протеаза, отнесенная к структурно-функциональному клану CA в БД MEROPS. Она обычно рассматривается как единая структура с убиквитин-подобным доменом Ubl2, который предположительно способствует связыванию протеазы с убиквитином [36]. Этот фермент расщепляет сайты Nsp1/Nsp2, Nsp2/Nsp3 и Nsp3/Nsp4 [37]. PLpro также отщепляет цепи убиквитина и ISG15 от хозяйских белков [37]. Ковалентное присоединение этих сравнительно небольших полипептидных цепей к модифицируемым белкам играет важную роль в реакциях врожденного иммунитета [38]. Такая специфичность обусловлена тем, что протеаза PLpro близка по структуре к деубиквитиназам семейства USP, экспрессируемым в том числе и у человека [39]. Поскольку деубиквитинирующие ферменты человека – это необходимый компонент регуляторных процессов, следует учитывать, что ингибирование PLpro может приводить к возникновению побочных эффектов.
У коронавирусов в ходе инфекционного процесса формируются репликативные органеллы, которые служат платформой для функционирования комплекса вирусных белков (Replication-transcription complex, RTC), осуществляющих репликацию и транскрипцию вирусных РНК. Репликативные органеллы представляют собой пузырьки, окруженные двойной мембраной (DMV), которые образуются из мембран эндоплазматического ретикулума (ЭПР) [40]. DMV экранируют возникающие при синтезе РНК двухцепочечные интермедиаты, которые распознаются рецепторами врожденного иммунитета [41].
Формирование DMV происходит при взаимодействии Nsp3 и Nsp4, имеющих в своем составе, соответственно, два и четыре трансмембранных участка [42]. Петли обоих белков, обращенные в просвет ЭПР, связываются между собой, инициируя искривление мембран ЭПР и отпочкование мембранных везикул (рисунок 5).
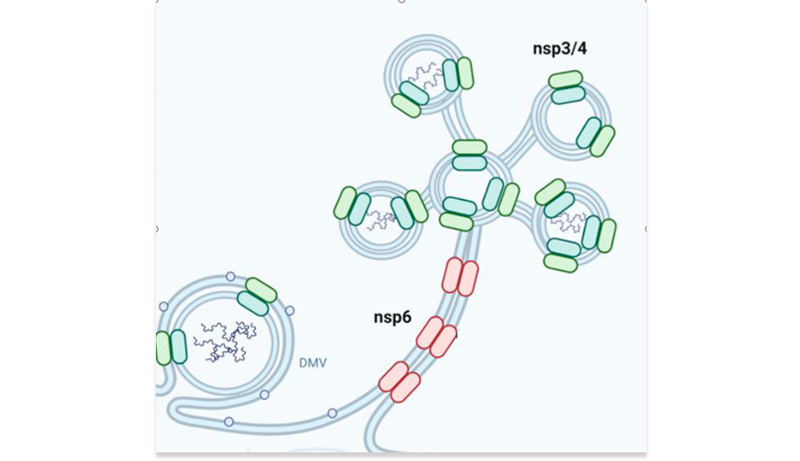
Процесс завершается путем стягивания мембран по принципу «застежки-молнии» за счет спаривания единиц белка Nsp6, каждая из которых заякорена в мембране ЭПР шестью спиралями [43].
С учетом описанного механизма представляется целесообразным поиск ингибиторов, нарушающих описанные взаимодействия. Потенциальные ингибиторы были предложены на основе виртуального скрининга, однако соответствующее экспериментальное тестирование не проведено [44].
Созревание DMV происходит при участии белков хозяина, необходимых для формирования аутофагосом - клеточных органелл с двойной мембраной [45]. Данные о влиянии Nsp6 на формирование аутофагосом также указывают на сходные механизмы формирования структур обоих типов [43].
Nsp3, будучи встроенным в двойную мембрану DMV посредством трансмембранных спиралей ТМ1 и ТМ2, образует поры, каждая из которых содержит по шесть субъединиц данного белка [46]. Эти каналы служат для транспорта геномных и субгеномных РНК между цитоплазмой и DMV, который осуществляется при взаимодействии Ubl1-домена Nsp3 c нуклеокапсидным белком N [47].
N-концевая область Nsp3 с доменами Ubl1 и Ubl2 на концах (рисунок 4), критически важна для процессов, которые запускают изменения в мембране ЭПР, необходимые для формирования DMV [48]. Этот участок включает в числе других три компонента cо сходной пространственной укладкой: Масro 1, Масro 2 и Масro 3.
Домен Macro 1 отщепляет от белков остатки AДФ-рибозы или поли-AДФ-рибозы, нарушая функции белков организма-хозяина, в том числе связанные с иммунитетом. SARS-CoV-2 c делециeй этого домена способен к репликации, но при этом высоко чувствителен к иммунным реакциям, опосредованным интерфероном гамма [49]. Macro 1 входит в надсемейство макродоменов, представленных в белках вирусов, бактерий и эукариот [50]. Два ингибитора Macro 1 были найдены путем виртуального скрининга на основе ингибиторов PARG (структурно сходного белка человека) и последующей кристаллизации в комплексе с мишенью [51]. Ряд ингибиторов Macro 1 был выявлен несколькими группами исследователей с помощью высокопроизводительного экспериментального скрининга [52].
Участки Macro 2 (SUD-N) и Macro 3 (SUD-M), сходны по структуре с Macro 1, но отличаются по функции. Вместе с субдоменом DPUP (SUD-C) они объединены в домен SUD, который, как полагают, взаимодействуя с факторами трансляции, способствует синтезу белков на вирусных мРНК [53]. Данные по связыванию SUB-N и SUB-C с олигогуаниновыми последовательностями РНК указывают на возможное участие SUD в работе репликативного комплекса [36].
Белок Nsp5 (главная протеаза, 3CLpro) расщепляет все протеолитические сайты в полипротеине, начиная с Nsp4/Nsp5 и заканчивая на Nsp15/Nsp16. Показано также, что 3CLpro расщепляет и хозяйские белки, в том числе и те, которые вовлечены в формирование реакций иммунного ответа [54-56]. 3CLpro активен в форме димера [57]. Протеазы указанного типа характеризуются трехмерной укладкой по типу химотрипсина. Несмотря на то, что значимое сходство последовательностей можно обнаружить только в пределах одного семейства, подобие трехмерных структур протеаз позволяет отнести их к одному надсемейству. Белки с таким типом пространственной укладки найдены у вирусов разных семейств, а также у эукариот, включая человека [58]. Согласно классификации MEROPS эти ферменты отнесены к структурно-функциональному клану PA.
Перспективное направление при поиске ингибиторов 3CLpro - разработка пептидомиметиков, имитирующих сайты протеолиза в белках-субстратах, с модификацией химических групп, связываемых карманами активного центра. На этой основе осуществлен молекулярный дизайн ингибитора нирматрелвир, внедренного в медицинскую практику в составе препарата паксловид [59].
PA-протеазы энтеро- и коронавирусов характеризуются сходными паттернами сайтов протеолиза в расщепляемых белках [60] (рисунок 6). Это служит доводом к репозиционированию ингибиторов протеаз, разработанных для других вирусов. Предложен ряд ингибиторов широкого спектра действия, которые подавляют репликацию как корона-, так и энтеровирусов [60]. Следует отметить что ранее был разработан ингибитор широкого спектра GC376, активный против норо-, корона- и пикорнавирусов (включают род энтеровирусов) как на уровне ферментативных тестов, так и клеточных моделей. Препарат показал эффективность при лечении перитонита кошек, вызываемого коронавирусом FIPV [61, 62].
При сопоставлении трехмерных структур с помощью метода DALI [63] было показано, что 3СLpro коронавирусов в большей степени сходны с протеазами энтеровирусов чем с протеазой NS3 вируса гепатита С (ВГС), которая также относится к надсемейству PA. Паттерны сайтов протеолиза, характерные для NS3 ВГС [64] существенно отличаются от паттернов, установленных для протеаз энтеро- и коронавирусов (рисунок 6). Этот пример указывает на перспективность структурных сопоставлений при репозиционировании ингибиторов вирусных ферментов.
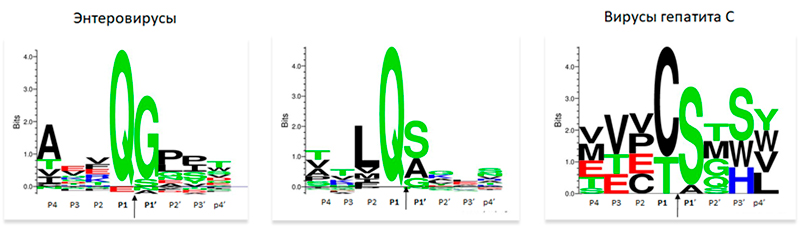
Для сайтов, расщепляемых протеазами человека, не характерен глютамин в позиции P1. Полагают что это должно гарантировать низкую вероятность побочных эффектов для ингибиторов 3CLpro [66]. Исследования ингибитора помотрелвира, действующего на активный сайт 3CLpro, показали его эффективность in vitro против протеаз SARS-CoV-2 и других коронавирусов вместе с высокой селективностью в отношении протеаз человека [67].
Хотя мутации 3CLpro достаточно редки, вероятность появления резистентных штаммов нельзя исключать при применении в клинике противовирусных препаратов. Недавние исследования по оценке устойчивых к нирматрелвиру вариантов SARS-CoV-2 были проведены на культурах клеток, инфицированных SARS-CoV-2 или химерным вирусом везикулярного стоматита [68-70]. В этих исследованиях серия пассажей с нирматрелвиром приводила к появлению устойчивых мутантных форм. Отмечены мутации, приводящие к снижению чувствительности к нирматрелвиру более чем в 100 раз [68, 70]. Большинство этих мутаций идентифицированы в последовательностях, депонированных в хранилищах данных, – в том числе поступивших до клинического применения комбинации нирматрелвира в составе препарата Paxlovid [69]. Систематическое исследование естественных вариантов 3CLpro SARS-CoV-2 позволило выявить 22 мутации, которые ассоциированы с более чем десятикратным повышением устойчивости к нирматрелвиру. Все эти замены были локализованы в пяти аминокислотных позициях кармана, связывающего ингибитор [71]. С целью преодоления резистентности продолжаются работы по конструированию новых ингибиторов 3CL на основе модификаций химических групп в пептидомиметиках [72].
Еще один подход к нейтрализаци протелитической активности 3CLpro основан на технологии PROTAC (PROteolysis TArgeting Chimeras), которая заключается в использовании химерной молекулы из двух компонентов - лиганда целевого белка и лиганда убиквитинлигазы Е3 [73]. Такая конструкция «притягивает» целевой белок к Е3, что приводит к убиквитинированию белка с последующей протеасомной деградацией. Использование соединений, сконструированных по указанному принципу, позволило значительно снизить уровень вирусных белков в клетках, инфицированных SARS-CoV-2 [74, 75].
Заслуживают внимания и исследования, направленные на поиск аллостерических ингибиторов, нарушающих димеризацию 3CLpro [76].
2.1 Репликативно-транскрипционный комплекс (RTC)
Коронавирусы имеют необычайно большой по сравнению с другими РНК-содержащими вирусами набор ферментов, связанных с воспроизводством и экспрессией вирусного генома [77]. Белки Nsp7, 8, 9, 10, 12, 13, 14, 15 и 16, собираются в RTC в составе репликативных органелл DMV (рисунок 7). В этом комплексе синтезируются новые копии РНК-генома и субгеномные матричные РНК структурных и вспомогательных белков [77].
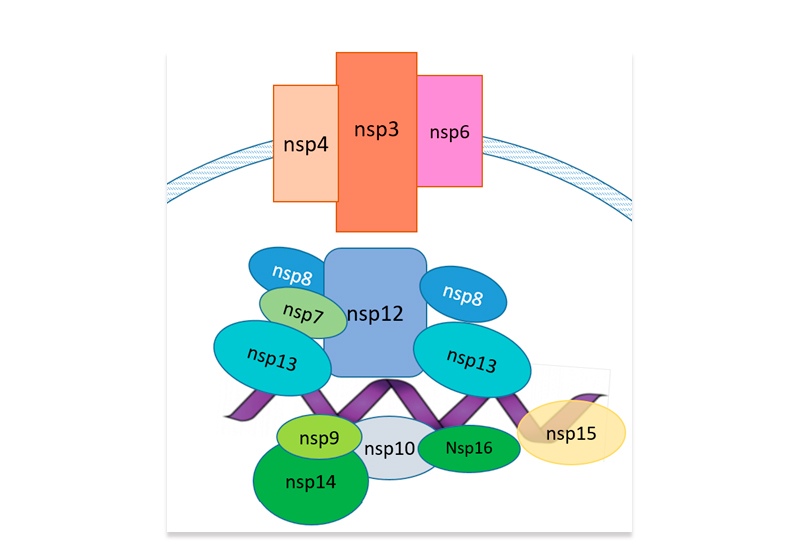
Белок Nsp9 участвует в инициировании синтеза РНК [78]. Результаты недавнего исследования также указывают на то, что Nsp9 участвует в инициации транскрипции в кооперации с хозяйским белком SND1 [79].
Nsp10 образует вместе с N-концевым доменом (ExoN) Nsp14 корректирующий комплекс (proofreading complex) и активирует метилтрансферазу Nsp16 [77]. Область генома, кодирующая этот белок, непосредственно предшествует сайту сдвига рамки (frameshifting). Связываясь с рибосомой, Nsp10 участвует в регуляции сдвига рамки, который необходим для синтеза полноразмерного полипротеина pp1ab и формирования полного набора неструктурных белков [28].
Белок Nsp12 включает домены NiRAN и RdRp, соединенные промежуточным Interface-доменом [80, 81] (рисунок 8).
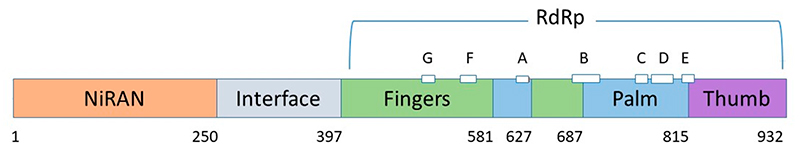
Домен NiRAN (около 250 остатков) присутствует в РНК-репликазах всех вирусов таксономического порядка Nidovirales. Он структурно сходен с протеинкиназами и проявляет фосфотрансферазную активность [82]. Этот домен участвует в формировании 5’кэпа [77]. Возможно также, что NiRAN обеспечивает ковалентное связывание между белком Nsp9 и монофосфорилированной РНК как промежуточный этап при синтезе 5’кэпа [83].
Домен RdRp (более 500 остатков) представляет собой РНК-зависимую РНК-полимеразу, которая в комплексе с Nsp7 и Nsp8 осуществляет репликацию геномной РНК и транскрипцию субгеномных РНК [80]. Этот фермент относится к большой группе структурно и функционально сходных белков (DNA/RNA polymerase superfamily), осуществляющих функцию нуклеотидной полимеразы у вирусов и ряда других биологических видов [84, 85].
Нуклеотидные полимеразы РНК-содержащих вирусов, включая обратные транскриптазы ретровирусов, характеризуются сходными паттернами на уровне аминокислотных последовательностей. Соответствующие мотивы A-G включают функционально значимые консервативные остатки [86]. Сопоставление трехмерных структур RdRp SARS-CoV-2 со структурами других вирусных протеаз позволило локализовать в ее последовательности те же мотивы, формирующие активный сайт фермента (рисунок 5) [80, 81].
Более близкое сходство аминокислотных последовательностей RdRp обнаруживается у вирусов из разных семейств с положительным одноцепочечным РНК-геномом (запись в PS50507 БД PROSITE).
Таким образом, для репозиционирования лекарственных препаратов, активных против RdRp других вирусов, открываются широкие возможности.
Нуклеозидные аналоги ингибиторов RdRp, такие как ремдесивир и молнупиравир, разрабатывались как средства, действенные против вирусов с РНК-геномом, таких как ВИЧ [87] или ВГС [88]. Их активность связана с нарушением синтеза РНК за счет конкуренции с естественными нуклеотидами за встраивание в растущую цепь [89]. Спустя 18 месяцев после начала пандемии было проведено исследование более чем пяти миллионов последовательностей SARS-CoV-2, депонированных в БД GISAID, на наличие мутаций, обуславливающих резистентность к ремдесивиру in vitro [90]. Несмотря на интенсивное клиническое применение препарата, частоты этих мутаций оказались крайне низки.
Указанное выше надсемейство структурно-подобных ДНК/РНК-полимераз включает 16 белков человека, депонированных в рецензируемой части БД UniProt. В их число входит митохондриальная РНК-полимераза человека (POLRMT), которая ингибируется рядом нуклеозидных аналогов [91]. Хотя воздействие ремдесивира на этот фермент не носит выраженного характера, оно рассматривается как одна из возможных причин нежелательных кардиотоксических эффектов [92].
В качестве более селективных средств исследуются ненуклеозидные ингибиторы RdRp, которые блокируют вход РНК, либо связываются с аллостерическими карманами в структуре полимеразы, препятствуя необходимым для связывания РНК конформационным изменениям и тем самым подавляя активность фермента [93, 94].
Белок Nsp13 функционирует как хеликаза с использованием АТФ, расплетая двуцепочечные РНК-интермедиаты. Также этот белок участвует в формировании 5’кэпа [77]. Nsp13 относится к структурно-функциональному надсемейству SF1 хеликаз [95], которые встречаются как у вирусов, так и у других видов, включая человека [96]. В RTC присутствуют две копии Nsp13, связанных с разными субдоменами RdRp.
N-концевой домен белка Nsp14 (ExoN) в комплексе с Nsp10 проявляет активность корректирующей экзорибонуклеазы (proofreading). Фермент в процессе синтеза РНК удаляет нуклеотиды, нарушающие комплементарность шаблонной и растущей цепей [97]. Благодаря этому механизму, некоторые нуклеозидные аналоги неэффективны при лечении коронавирусных инфекций [77]. Виртуальный скрининг библиотеки нуклеозидных аналогов с последующей экспериментальной проверкой выявил перспективные соединения, ингибирующие как RdRp, так и ExoN и, таким образом подавляющие репликацию вируса [98].
С-концевой домен Nsp14 (N7-MTase) работает как метилтрансфераза при формировании 5’кэпа [77].
Белок Nsp15 (уридилат-специфичная эндорибонуклеаза, NendoU) препятствует развитию иммунных реакций. Как показано для SARS-CoV, этот фермент укорачивает полиуридиновые участки на 5’-концах минус-цепей, лимитируя уровень РНК-интермедиатов, которые служат сигналами для запуска реакций врожденного иммунитета [99].
Белок Nsp16 (2'O-метилтрансфераза, 2′-O-MTase), в сочетании с Nsp10, осуществляет второе метилирование 5’кэпа [77].
Формирование 5’-кэпа – важный этап при синтезе вирусной РНК. Эта группа необходима для поддержания стабильности РНК, запуска трансляции и защиты от клеточных экзонуклеаз [83]. Присоединение 5’-кэпа происходит в несколько этапов [77]. Nsp13 отщепляет гамма-фосфат от 5’-конца РНК. NiRAN-домен из Nsp12 присоединяет к РНК группу гуанозин-монофосфата. В завершение 5’-кэп метилируется метилтрансферазой Nsp14 (доменом N7-MTase), а затем метилируется Nsp16 (2′-O-MTase).
3. Структурные белки и сборка вириона. Высвобождение из клетки.
Субгеномные рамки считывания кодируют четыре структурных белка S, N, M и E, образующие вирусную частицу.
S-белок, участвующий в самом первом этапе жизненного цикла вируса охарактеризован ранее.
N-белок (419 аминокислотных остатков) формирует нуклеокапсид в форме спирали, которая содержит одну цепь геномной РНК и более 4000 субъединиц N-белка [100].
N-белок содержит два структурированных домена NTD и CTD, а также три неупорядоченных участка на N-конце (N-arm), между доменами (LKR) и на C-конце (C-tail). Основные остатки NTD-домена и других частей молекулы белка обуславливают ее связывание с РНК [101]. N-концевая часть CTD-домена также отвечает также за связывание РНК за счет бороздки, заполненной основными остатками [102], тогда как С-концевая часть – за димеризацию. Димеры N-белка укладываются в надструктуры более высокого порядка. В составе LKR выявлен участок SR, который связывается с Ubl1-доменом Nsp3, при взаимодействии N-белка с комплексом RTC [47], что обеспечивает обмен РНК между репликативными органеллами и цитоплазмой.
N-белок может перехватывать двухцепочечные РНК-интермедиаты, препятствуя развитию защитных реакций, таких как антивирусная РНК интерференция [103].
Несмотря на то, что нуклеопротеин локализован внутри вириона, антительный ответ на него выражен сильнее и развивается раньше в сравнении с поверхностным S-белком. Соответствующие серологические тесты помогают быстро оценить особенности инфекционного процесса [104]. Кроме того, N-белок рассматривается как стимулятор стабильного клеточного иммунитета [105]. Сочетание S- и N-белков в вакцинных конструкциях представляется перспективным направлением [106]. На основе генноинженерной конструкции, содержащей полноразмерный N-белок, а также фрагменты белков M, S и Е, отечественными учеными создан инновационный препарат КоронаДерм-PS для оценки Т-клеточного иммунитета при коронавирусной инфекции с высокой чувствительностью. Отобранные участки структурных белков включают высоко консервативные эпитопы среди различных штаммов SARS-CoV-2, что обеспечивает надежность диагностики. Безопасность и эффективность КоронаДерм-PS продемонстрированы в ходе слепого плацебо-контролируемого клинического исследования [107].
Мембранный белок (M-белок) содержит 222 аминокислотных остатка и формирует димер. Он играет ключевую роль в сборке и морфогенезе вирусных частиц [108]. N-концевой домен М-белка встроен в мембрану. C-концевой домен М-белка обращен в цитоплазму и в конечном счете локализуется внутри собранного вириона. Взаимодействие M-белка со структурными белками N, E и S необходимо для сборки вириона [109]. Эти мембранные белки после синтеза располагаются рядом в мембране ЭПР (рисунок 2) и мигрируют к месту сборки в промежуточном компартменте ERGIC. За счет взаимодействия между С-концевым доменом М-белка и нуклеокапсидом, последний “подтягивается” к месту сборки [108, 109]. Мембрана замыкается вокруг вириона, который поступает в аппарат Гольджи. Далее вирион включается в лизосому и покидает клетку путем экзоцитоза.
Оболочечный белок E включает 75 аминокислотных остатков. Центральный гидрофобный участок образует трансмембранную спираль. Эти спирали собираются в пентамер, действующий как ионный канал [110]. Таким образом, E-белок – типичный представитель семейства виропоринов, которые необходимы для сборки вирионов и их освобождения из клетки [111]. Блокаторы ионных каналов рассматриваются как потенциальные противовирусные препараты [112]. На примере других коронавирусов показано, что большинство молекул E-белка локализованы не в оболочке вируса, а в ЭПР, аппарате Гольджи и ERGIC – то есть в тех структурах, которые вовлечены в сборку и отпочкование вирионов [110].
4. Вспомогательные белки
Эти белки кодируются рамками (ORF) 3a, 3b, 3c, 3d, 6, 7a, 7b, 8, 9b, 9с и 10. Они не участвуют в репликации вируса, однако вовлечены в патогенез, включая защиту вируса от иммунных реакций [9]. Экспериментальные результаты показывают, что вспомогательные белки ингибируют продукцию интерферона, способствуют обострению воспалительных процессов, индуцируют неконтролируемый апоптоз, снижают антигенную презентацию, блокируют аутофагию, провоцируют стресс ЭПР [10].
Показано, что мутации, выключающие некоторые вспомогательные гены, некритичны для репликации SARS-CoV-2 [113] из БД GISAID. У пациентов, инфицированных вирусом с делецией, затрагивающей три неструктурных белка ORF7a, ORF7b и ORF8, отмечено сокращение сроков госпитализации. При этом, тяжесть заболевания и выраженность иммунного ответа были такими же, как у пациентов, зараженных вирусом “дикого” типа [114].
Самый большой вспомогательный белок ORF3a взаимодействует с белками клеточного транспорта и, таким образом, вероятно, вовлекается в процессы вирусного высвобождения и уклонения от иммунного ответа [115]. Делеция гена ORF3a приводит к значительному угнетению воспроизводства вируса в зараженных клетках [116].
5. Изменчивость белков SARS-CoV-2
Изменчивость белков вируса в ходе эпидемического процесса важна при разработке лекарств и вакцин. Данные по динамике мутаций у SARS-CoV-2 опубликованы в работе [117].
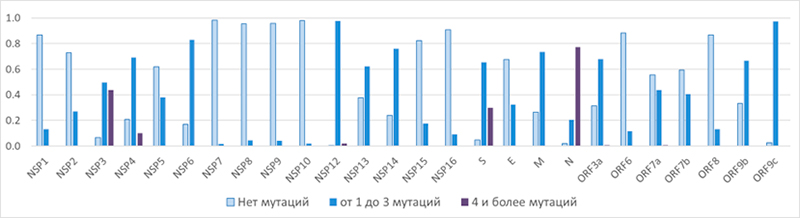
Вполне понятна высокая изменчивость S-белка как как мишени нейтрализующих антител. Такая популярная лекарственная мишень как Nsp5 (3CLpro) относительно консервативна, а Nsp12 (RdRp) более изменчива.
6. Белки человека как возможные мишени
С начала пандемии ковид 19 значительные усилия были направлены на разработку лекарств, действующих на белки человека (хозяйские мишени), непосредственно взаимодействующие с белками вируса или включенные в патогенез. Прежде всего стоит упомянуть о мишенях, для которых установлены, крайней мере in vitro, лиганды с противовирусным эффектом.
Блокада ACE2 нарушает взаимодействие этого рецептора с S-белком и, тем самым, перекрывает основной путь для входа вируса [18, 21].
Протеаза фурин расщепляет полипептид S по сайту S1/S2, вероятно, еще до выхода вириона из клетки [12], что способствует формированию функционального комплекса S1-S2 [118].
Трансмембранные сериновые протеазы (TTSPs), включая TMPRSS2, и катепсин L необходимы для активации пептида слияния путем расщепления S2-белка по сайту S2’. TTSPs действуют на поверхности клетки, а катепсин L в эндосоме. В обоих случаях расщепление белка S2 сопровождается структурной перестройкой S-белка, слиянием мембран и поступлением вирусного материала в цитоплазму [12, 13].
Лиганды рецептора сигма-1 (σ1R) часто идентифицируют при скрининге лекарств in vitro, направленном на выявление соединений, активных против коронавирусов [119]. σ1R – лиганд-управляемый шаперон, модулирующий процессы, связанные со стрессом эндоплазматического ретикулума – явления, которое развивается, в том числе, и при коронавирусной инфекции. Последняя, как уже отмечалось, сопровождается перестройкой внутренних мембран клетки. Показано взаимодействие σ1R с вирусным белком Nsp6, участвующим в формировании DMV [120].
Один из подходов к поиску хозяйских мишеней основан на выявлении белков человека, сходных с вирусными белками, путем трехмерного сопоставления [121]. Для определенных таким образом белков человека извлекаются установленные данные по белок-белковым взаимодействиям. Это позволяет наметить вероятные хозяйские мишени, которые могут взаимодействовать с белками вируса.
Сегодня ни один из препаратов, действующих на хозяйские мишени, группы не получил одобрения. Это связано, в том числе, с возможными побочными эффектами и с наличием дублирующих путей в жизненном цикле вируса [122].
Заключение
На данный момент вирусные мишени остаются ключевым предметом исследования при поиске соединений, активных против вновь выявленного патогенного вируса [122].
Сделан вывод, что при выборе лекарственных мишеней нового вируса следует ориентироваться на данные по белкам других вирусов, сходным с этими мишенями, и при этом уделять особое внимание структуре лиганд-связывающих карманов. Касательно этих белков следует учитывать сведения по активных против них лигандам, включая результаты тестирования in vitro и in vivo с четким пониманием механизма действия, а также результаты клинических испытаний и опыт терапевтического применения [122]. Этим критериям в наибольшей степени отвечают 3CLpro (nsp5) и RdRp (nsp12).
При разработке лекарственных средств, как на основе репозиционированных соединений, так и de novo, обращают внимание на структурные особенности белков-мишеней, обеспечивающих субстратную и ингибиторную специфичность. Это наглядно показано на примере протеаз энтеро- и коронавирусов, характеризующихся близким сходством как по трехмерной структуре, так и по паттернам протеолиза.
Ряд белков и белковых доменов SARS-CoV-2, таких как 3CLpro, PLpro, RdRp, Macro 1 и др., сходны с белками человека на уровне трехмерной организации и отнесены к соответствующим структурно-функциональные надсемействам. В связи с этим необходимо исследовать селективность ингибиторов для исключения или минимизации побочных эффектов. С другой стороны, такое сходство положено в основу поиска ингибиторов, блокирующих взаимодействие белков человека и вируса [121].
Сложности при разработке лекарств против COVID-19 связаны и с тем, что SARS-CoV-2 содержит довольно большой арсенал белков, которые, взаимодействуя между собой и белками человека, обеспечивают не только реализацию жизненного цикла вируса, но и защиту от реакций врожденного иммунитета.
Примером комплексного подхода к поиску эффективных антикоронавирусных соединений является изучение ингибиторов двойного действия против РНК-репликазы RdRp и корректирующей экзонуклеазы ExoN [98].
Цитируемая литература:
1. Wu F. et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265-269. DOI: 10.1038/s41586-020-2008-3
2. Finkel Y. et al. The coding capacity of SARS-CoV-2. Nature. 2021;589(7840):125-130. DOI: 10.1038/s41586-020-2739-1
3. Bai C., Zhong Q., Gao G.F. Overview of SARS-CoV-2 genome-encoded proteins. Sci. China Life Sci. 2022;65(2):280-294. DOI: 10.1007/s11427-021-1964-4
4. Justo Arevalo S. et al. What do we know about the function of SARS-CoV-2 proteins? Front Immunol. 2023;14:1249607. DOI: 10.3389/fimmu.2023.1249607
5. Jungreis I. et al. Conflicting and ambiguous names of overlapping ORFs in the SARS-CoV-2 genome: A homology-based resolution. Virology. 2021;558:145-151. DOI: 10.1016/j.virol.2021.02.013
6. Tam D. et al. Targeting SARS-CoV-2 non-structural proteins. Int J Mol Sci. 2023;24(16):13002. DOI: 10.3390/ijms241613002
7. Yao H. Song Y., Chen Y. at al. Molecular architecture of the SARS-CoV-2 virus. Cell. 2020;183(3):730-738. DOI: 10.1016/j.cell.2020.09.018
8. Hadi J., Dunowska M., Wu S., Brightwell G. Control Measures for SARS-CoV-2: A Review on Light-Based Inactivation of Single-Stranded RNA Viruses. Pathogens. 2020 Sep 8;9(9):737. DOI: 10.3390/pathogens9090737
9. Zandi M. et al. The role of SARS-CoV-2 accessory proteins in immune evasion. Biomed Pharmacother. 2022;156:113889. DOI: 10.1016/j.biopha.2022.113889
10. Hurtado-Tamayo J. et al. Contribution to pathogenesis of accessory proteins of deadly human coronaviruses. Front Cell Infect Microbiol. 2023; 13:1166839. DOI: 10.3389/fcimb.2023.1166839
11. Zhao L., Li S., Zhong W. Mechanism of action of small-molecule agents in ongoing clinical trials for SARS-CoV-2. 2022;13:840639. DOI: 10.3389/fphar.2022.840639
12. Jackson C.B. et al. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol. 2022;23(1):3-20. DOI: 10.1038/s41580-021-00418-x
13. Zabiegala A., Kim Y., Chang K.O. Roles of host proteases in the entry of SARS-CoV-2. Anim Dis. 2023;3(1):12. DOI: 10.1186/s44149-023-00075-x
14. Gudowska-Sawczuk M., Mroczko B. The role of neuropilin-1 (NRP-1) in SARS-CoV-2 infection: Review. J Clin Med. 2021;10(13):2772. DOI: 10.3390/jcm10132772
15. Fenizia C. et al. SARS-CoV-2 Entry: At the Crossroads of CD147 and ACE2. Cells. 2021;10(6):1434. DOI: 10.3390/cells10061434
16. Lempp F.A. et al. Lectins enhance SARS-CoV-2 infection and influence neutralizing antibodies. Nature. 2021;598(7880):342-347. DOI: 10.1038/s41586-021-03925-1
17. Mori Y. et al. KIM-1/TIM-1 is a receptor for SARS-CoV-2 in lung and kidney. medRxiv.Preprint. 2022 DOI: 10.1101/2020.09.16.20190694
18. Lim S.P. Targeting SARS-CoV-2 and host cell receptor interactions. Antiviral Res. 2023;210:105514. DOI: 10.1016/j.antiviral.2022.105514
19. Xia X. Identification of host receptors for viral entry and beyond: a perspective from the spike of SARS-CoV-2. Front Microbiol. 2023;14:1188249. DOI: 10.3389/fmicb.2023.1188249
20. Shilts J. et al. No evidence for basigin/CD147 as a direct SARS-CoV-2 spike binding receptor. Sci Rep. 2021;11(1):413. DOI: 10.1038/s41598-020-80464-1
21. Zhang H. et al. Advances in developing ACE2 derivatives against SARS-CoV-2. Lancet Microbe. 2023;4(5):e369-e378. DOI: 10.1016/s2666-5247(23)00011-3
22. Parisi G. et al. Design of protein-binding peptides with controlled binding affinity: the case of SARS-CoV-2 receptor binding domain and angiotensin-converting enzyme 2 derived peptides. Front Mol Biosci. 2024;10:1332359. DOI: 10.3389/fmolb.2023.1332359
23. Ullah A. Identification of new pharmacophore against SARS-CoV-2 spike protein by multi-fold computational and biochemical techniques. Sci Rep. 2024; 14(1):3590. DOI: 10.1038/s41598-024-53911-6
24. Guo L. et al. Targetable elements in SARS-CoV-2 S2 subunit for the design of pan-coronavirus fusion inhibitors and vaccines. Signal Transduct Target Ther. 2023; 8(1):197. DOI: 10.1038/s41392-023-01472-x
25. Li X. et al. Spike protein mediated membrane fusion during SARS-CoV-2 infection. J Med Virol. 2023;95(1):e28212. DOI: 10.1002/jmv.28212
26. Maeda Y. Differential ability of spike protein of SARS-CoV-2 variants to downregulate ACE2. Int J Mol Sci. 2024;25(2):1353. DOI: 10.3390/ijms25021353
27. Long S. SARS-CoV-2 subgenomic RNAs: characterization, utility, and perspectives. Viruses. 2021; 13(10):1923. DOI: 10.3390/v13101923
28. Bhatt P.R. et al. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. Science. 2021;372(6548):1306-1313. DOI: 10.1126/science.abf3546
29. Simeoni M. et al. I(nsp1)ecting SARS-CoV-2-ribosome interactions. Commun Biol. 2021;4(1):715. DOI: 10.1038/s42003-021-02265-0
30. Tardivat Y. et al. SARS-CoV-2 NSP1 induces mRNA cleavages on the ribosome. Nucleic Acids Res. 2023;51(16):8677-8690. DOI: 10.1093/nar/gkad627
31. Zhang K. et al. Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci Adv. 2021;7(6):eabe7386. DOI: 10.1126/sciadv.abe7386
32. Kilkenny M.L. et al. Structural basis for the interaction of SARS-CoV-2 virulence factor nsp1 with DNA polymerase α-primase. Protein Sci. 2022;31(2):333-344. DOI: 10.1002/pro.4220
33. Graham R.L. et al. The nsp2 replicase proteins of murine hepatitis virus and severe acute respiratory syndrome coronavirus are dispensable for viral replication. J Virol. 2005;79(21):13399-13411. DOI: 10.1007/978-0-387-33012-9_10
34. Gupta M. et al. CryoEM and AI reveal a structure of SARS-CoV-2 Nsp2, a multifunctional protein involved in key host processes. bioRxiv.Preprint. 2021; DOI: 10.1101/2021.05.10.443524
35. Naeli P. et al. The SARS-CoV-2 protein nsp2 enhances microRNA-mediated translational repression. J Cell Sci. 2023;136(19):jcs261286. DOI: 10.1242/jcs.261286
36. Lei J., Kusov Y., Hilgenfeld R. Nsp3 of coronaviruses: structures and functions of a large multi-domain protein. Antiviral Res. 2018;149:58-74. DOI: 10.1016/j.antiviral.2017.11.001
37. Tan H. et al. Progress and challenges in targeting the SARS-CoV-2 papain-like protease. J Med Chem. 2022;65(11):7561-7580. DOI: 10.1021/acs.jmedchem.2c00303
38. Shin D. et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587(7835):657-662. DOI: 10.1016/j.tim.2020.05.009
39. Osipiuk J. et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat Commun. 2021;12(1):743. DOI: 10.1038/s41467-021-21060-3
40. Wolff G. et al. Double-membrane vesicles as platforms for viral replication. Trends Microbiol. 2020;28(12):1022-1033. DOI: 10.1016/j.cplett.2021.138819
41. Thoresen D. et al. The molecular mechanism of RIG-I activation and signaling. Immunol Rev. 2021;304(1):154-168. DOI: 10.1111/imr.13022
42. Klatte N. et al. Modelling the transitioning of SARS-CoV-2 nsp3 and nsp4 lumenal regions towards a more stable state on complex formation. Int J Mol Sci. 2022;24(1):720. DOI: 10.3390/ijms24010720
43. Bills C., Xie X., Shi P.Y. The multiple roles of nsp6 in the molecular pathogenesis of SARS-CoV-2. Antiviral Res. 2023;213:105590. DOI: 10.1016/j.antiviral.2023.105590
44. Chakraborty J, Maity A, Sarkar H. A systematic drug repurposing approach to identify promising inhibitors from FDA-approved drugs against nsp4 protein of SARS-CoV-2. J Biomol Struct Dyn. 2023;41(2):550-559. DOI: 10.1080/07391102.2021.2009033
45. Ji M. et al. DMV biogenesis during β-coronavirus infection requires autophagy proteins VMP1 and TMEM41B. Autophagy. 2023;19(2):737-738. DOI: 10.1080/15548627.2022.2103783
46. Wolff G. et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science. 2020;369(6509):1395-1398. DOI: 10.1126/science.abd3629
47. Koetzner C.A. et al. Analysis of a crucial interaction between the coronavirus nucleocapsid protein and the major membrane-bound subunit of the viral replicase-transcriptase complex. Virology. 2022;567:1-14. DOI: 10.1016/j.virol.2021.12.004
48. Zimmermann L. et al. SARS-CoV-2 nsp3-4 suffice to form a pore shaping replication organelles. bioRxiv.Preprint. 2022; DOI: 10.1101/2022.10.21.513196
49. Alhammad Y.M. et al. SARS-CoV-2 Mac1 is required for IFN antagonism and efficient virus replication in cell culture and in mice. Proc Natl Acad Sci USA. 2023;120(35):e2302083120. DOI: 10.1101/2023.04.06.535927
50. Rack J.G., Perina D., Ahel I. Macrodomains: Structure, Function, Evolution, and Catalytic Activities. Annu Rev Biochem. 2016;85:431-54. DOI: 10.1146/annurev-biochem-060815-014935
51. Brosey C.A. et al. Targeting SARS-CoV-2 Nsp3 macrodomain structure with insights from human poly(ADP-ribose) glycohydrolase (PARG) structures with inhibitors. Prog Biophys Mol Biol. 2021;163:171-186. DOI: 10.1016/j.pbiomolbio.2021.02.002
52. O'Connor J.J., Ferraris D., Fehr A.R. An update on the current state of SARS-CoV-2 Mac1 inhibitors. Pathogens. 2023;12(10):1221. DOI: 10.3390/pathogens12101221
53. Li P. et al. Nsp3-N interactions are critical for SARS-CoV-2 fitness and virulence. Proc Natl Acad Sci USA. 2023;120(31):e2305674120. DOI: 10.1073/pnas.2305674120
54. Song L. et al. The main protease of SARS-CoV-2 cleaves histone deacetylases and DCP1A, attenuating the immune defense of the interferon-stimulated genes. J Biol Chem. 2023;299(3):102990. DOI: 10.1016/j.jbc.2023.102990
55. Wu Y. et al. Broad antagonism of coronaviruses nsp5 to evade the host antiviral responses by cleaving POLDIP3. PLoS Pathog. 2023;19(10):e1011702. DOI: 10.1371/journal.ppat.1011702
56. Ju X. et al. SARS-CoV-2 main protease cleaves MAGED2 to antagonize host antiviral defense. mBio. 2023;14(4):e0137323. DOI: 10.1128/mbio.01373-23
57. Parmar M. et al. Structural differences in 3C-like protease (Mpro) from SARS-CoV and SARS-CoV-2: molecular insights revealed by molecular dynamics simulations. Struct Chem. 2022;23:1-18. DOI: 10.1007/s11224-022-02089-6
58. Mönttinen H.A.M., Ravantti J.J., Poranen M.M. Structural comparison strengthens the higher-order classification of proteases related to chymotrypsin. PLoS One. 2019;14(5):e0216659. DOI: 10.1371/journal.pone.0216659
59. Marzi M., Vakil M.K., Bahmanyar M., Zarenezhad E. Paxlovid: mechanism of action, synthesis, and in silico study. Biomed Res. 2022 Int. V. 2022, Article ID 7341493. DOI: 10.1155/2022/7341493
60. Zhang L. et al. α-ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J Med Chem. 2020;63(9):4562-4578. DOI: 10.1021/acs.jmedchem.9b01828
61. Kim Y, et al. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J Virol. 2012;86(21):11754-11762. DOI: 10.1016/j.cplett.2021.138819
62. Kim Y. et al. Reversal of the Progression of Fatal Coronavirus Infection in Cats by a Broad-Spectrum Coronavirus Protease Inhibitor. PLoS Pathog. 2016;12(3):e1005531. DOI: 10.1371/journal.ppat.1005531
63. Holm L. Dali server: structural unification of protein families. Nucleic Acids Res. 2022;50(W1):W210-W215. DOI: 10.1093/nar/gkac387
64. Shiryaev S.A. et al. New details of HCV NS3/4A proteinase functionality revealed by a high-throughput cleavage assay. PLoS One. 2012;7(4):e35759. DOI: 10.1371/journal.pone.0035759
65. Thomsen M.C., Nielsen M. Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res. 2012;40(Web Server issue):W281-287. DOI: 10.1093/nar/gks469
66. Rut W. et al. SARS-CoV-2 Mpro inhibitors and activity-based probes for patient-sample imaging. Nat Chem Biol. 2021;17(2):222-228. DOI: 10.1038/s41589-020-00689-z
67. Tong X. et al. Evaluation of in vitro antiviral activity of SARS-CoV-2 Mpro inhibitor pomotrelvir and cross-resistance to nirmatrelvir resistance substitutions. Antimicrob Agents Chemother. 2023; 67(11):e0084023. DOI: 10.1128/aac.00840-23
68. Zhou Y. et al. Nirmatrelvir-resistant SARS-CoV-2 variants with high fitness in an infectious cell culture system. Sci Adv. 2022;8(51):eadd7197. DOI: 10.1126/sciadv.add7197
69. Heilmann E. et al. SARS-CoV-2 3CLpro mutations selected in a VSV-based system confer resistance to nirmatrelvir, ensitrelvir, and GC376. Sci Transl Med. 2023;15(678):eabq7360. DOI: 10.1126/scitranslmed.abq7360
70. Iketani S. et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature. 2023;613(7944):558-564. DOI: 10.1038/s41586-022-05514-2
71. Hu Y., Lewandowski E.M., Tan H. Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to Nirmatrelvir. ACS Cent Sci. 2023;9(8):1658-1669. DOI: 10.1021/acscentsci.3c00538
72. Schillings J. et al. A computational comparison between pomotrelvir and nirmatrelvir binding and reactivity with SARS-CoV-2 main protease. Implications for resistance mechanisms. ChemRxiv. 2024. DOI: 10.26434/chemrxiv-2024-thvcv
73. Ahmad H., Zia B., Husain H., Husain A. Recent advances in PROTAC-based antiviral strategies. Vaccines (Basel). 2023;11(2):270. DOI: 10.3390/vaccines11020270
74. Alugubelli Y.R. et al. Discovery of first-in-class PROTAC degraders of SARS-CoV-2 main protease. bioRxiv.Preprint. 2023; DOI: 10.1101/2023.09.29.560163
75. Grifagni D. et al. Development of a GC-376 Based Peptidomimetic PROTAC as a Degrader of 3-Chymotrypsin-like Protease of SARS-CoV-2. ACS Med Chem Lett. 2024;15(2):250-257. DOI: 10.1021/acsmedchemlett.3c00498
76. Giri-Rachman E.A. et al. The SARS-CoV-2 Mpro dimer-based screening system: a synthetic biology tool for identifying compounds with dimerization inhibitory potential. ACS Synth Biol. 2024;13(2):509-520. DOI: 10.1021/acssynbio.3c00446
77. Malone B. et al. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat Rev Mol Cell Biol. 2022;23(1):21-39. DOI: 10.1038/s41580-021-00432-z
78. Wang B., Svetlov D., Artsimovitch I. NMPylation and de-NMPylation of SARS-CoV-2 nsp9 by the NiRAN domain. Nucleic Acids Res. 2021;49(15):8822-8835. DOI: 10.1093/nar/gkab677
79. Schmidt N. et al. SND1 binds SARS-CoV-2 negative-sense RNA and promotes viral RNA synthesis through NSP9. Cell. 2023;186(22):4834-4850. DOI: 10.1016/j.cell.2023.09.002
80. Gao Y. et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. 2020; 368(6492):779-782. DOI: 10.1126/science.abb7498
81. Itoh S.G., Tanimoto S., Okumura H. Dynamic properties of SARS-CoV and SARS-CoV-2 RNA-dependent RNA polymerases studied by molecular dynamics simulations. Chem. Phys. Lett. 2021;778:138819. DOI: 10.1016/j.cplett.2021.138819
82. Dwivedy A. et al. Characterization of the NiRAN domain from RNA-dependent RNA polymerase provides insights into a potential therapeutic target against SARS-CoV-2. PLoS Comput Biol. 2021;17(9):e1009384. DOI: 10.1371/journal.pcbi.1009384
83. Park G.J. et al. The mechanism of RNA capping by SARS-CoV-2. Nature. 2022;609(7928):793-800. DOI: 10.1038/s41586-022-05185-z
84. Delarue M. et al. An attempt to unify the structure of polymerases. Protein Eng. 1990;3(6):461-467. DOI: 10.1093/protein/3.6.461
85. Hansen J.L., Long A.M., Schultz S.C. Structure of the RNA-dependent RNA polymerase of poliovirus. Structure. 1997;5(8):1109-1122. DOI: 10.1016/s0969-2126(97)00261-x
86. te Velthuis A.J. Common and unique features of viral RNA-dependent polymerases. Cell. Mol. Life Sci. 2014;71(22):4403-4420. DOI: 10.1007/s00018-014-1695-z
87. Amblard F. et al. HIV nucleoside reverse transcriptase inhibitors. Eur J Med Chem. 2022;240:114554. DOI: 10.1016/j.ejmech.2022.114554
88. Elfiky A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020; 248:117477. DOI: 10.1016/j.lfs.2020.117477
89. Comunale B.A. et al. The RNA-Dependent RNA functional implications of broad spectrum bioactive compounds targeting polymerase (RdRp) in the context of the COVID-19 pandemic. Viruses. 2023;15(12):2316. DOI: 10.3390/v15122316
90. Focosi D. et al. Very low levels of remdesivir resistance in SARS-COV-2 genomes after 18 months of massive usage during the COVID19 pandemic: A GISAID exploratory analysis. Antiviral Res. 2022;198:105247. DOI: 10.1016/j.antiviral.2022.105247
91. Jin Z. et al. Structure-activity relationship analysis of mitochondrial toxicity caused by antiviral ribonucleoside analogs. Antiviral Res. 2017;143:151-161. DOI: 10.1016/j.antiviral.2017.04.005
92. Kanagala S.G. et al. Remdesivir-induced bradycardia. South Med J. 2023;116(3):317-320. DOI: 10.14423/smj.0000000000001519
93. Brindani N. et al. Design, synthesis, docking, and biochemical characterization of non-nucleoside SARS-CoV-2 RdRp inhibitors. Bioorg Med Chem. 2023; 80:117179. DOI: 10.1016/j.bmc.2023.117179
94. Zhou B. et al. Recent advancements in the discovery of small-molecule non-nucleoside inhibitors targeting SARS-CoV-2 RdRp. Biomed Pharmacother. 2024;171:116180. DOI: 10.1016/j.biopha.2024.116180
95. Weber R., McCullagh M. Role of ATP in the RNA translocation mechanism of SARS-CoV-2 nsp13 helicase. J Phys Chem B. 2021;125(31):8787-8796. DOI: 10.1021/acs.jpcb.1c04528
96. Fairman-Williams M.E., Guenther U.P., Jankowsky E. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol. 2010;20(3):313-324. DOI: 10.1016/j.sbi.2010.03.011
97. Deval J., 4Gurard-Levin Z.A. Opportunities and challenges in targeting the proofreading activity of SARS-CoV-2 Polymerase Complex. Molecules. 2022;27(9):2918. DOI: 10.3390/molecules27092918
98. Rabie A.M., Abdalla M. Evaluation of a series of nucleoside analogs as effective anticoronaviral-2 drugs against the Omicron-B.1.1.529/BA.2 subvariant: a repurposing research study. Med Chem Res. 2023;32(2):326-341. DOI: 10.1007/s00044-022-02970-3
99. Hackbart M., Deng X., Baker S.C. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc Natl Acad Sci USA. 2020; 117(14):8094-8103. DOI: 10.1073/pnas.1921485117
100. Chechetkin V.R., Lobzin V.V. On the number of nucleoproteins in the assembly of coronaviruses: Consequences for COVID-19. arXiv.Preprint. 2022 DOI: 10.48550/arXiv.2210.17257
101. Bai Z. et al. The SARS-CoV-2 Nucleocapsid protein and its role in viral structure, biological functions, and a potential target for drug or vaccine mitigation. Viruses. 2021;13(6):1115. DOI: 10.3390/v13061115
102. Zinzula L. et al. High-resolution structure and biophysical characterization of the nucleocapsid phosphoprotein dimerization domain from the Covid-19 severe acute respiratory syndrome coronavirus 2. Biochem Biophys Res Commun. 2021;538:54-62. DOI: 10.1016/j.bbrc.2020.09.131
103. Mu J. et al. SARS-CoV-2-encoded nucleocapsid protein acts as a viral suppressor of RNA interference in cells. Sci China Life Sci. 2020;63(9):1413-1416. DOI: 10.1007/s11427-020-1692-1
104. Kim Y. et al. Epitopes recognition of SARS-CoV-2 nucleocapsid RNA binding domain by human monoclonal antibodies. iScience. 2024;27(2):108976. DOI: 10.1016/j.isci.2024.108976
105. Choy C. et al. SARS-CoV-2 infection establishes a stable and age-independent CD8+ T cell response against a dominant nucleocapsid epitope using restricted T cell receptors. Nat Commun. 2023;14(1):6725. DOI: 10.1038/s41467-023-42430-z
106. Song W. et al. The role of SARS-CoV-2 N protein in diagnosis and vaccination in the context of emerging variants: present status and prospects. Front Microbiol. 2023;14:1217567. DOI: 10.3389/fmicb.2023.1217567
107. Симбирцев А.С. Разработка инновационного медицинского препарата для оценки Т-клеточного иммунитета при коронавирусной инфекции. Доклад на Заседании Бюро Секции медико-биологических наук Отделения медицинских наук РАН 28 ноября 2023 года.
108. Zhang Z. et al. Structure of SARS-CoV-2 membrane protein essential for virus assembly. Nat Commun. 2022;13(1):4399. DOI: 10.1038/s41467-022-32019-3
109. Kung Y.A. et al. Molecular virology of SARS-CoV-2 and related coronaviruses. Microbiol Mol Biol Rev. 2022;86(2):e0002621. DOI: 10.1128/mmbr.00026-21
110. Cao Y. et al. Characterization of the SARS-CoV-2 E protein: sequence, structure, viroporin, and inhibitors. Protein Sci. 2021;30(6):1114-1130. DOI: 10.1002/pro.4075
111. Nieva J., Madan V., Carrasco L. Viroporins: structure and biological functions. Nat Rev Microbiol. 2012;10(8):563–574. DOI: 10.1038/nrmicro2820
112. Breitinger U. et al. Patch-clamp studies and cell viability assays suggest a distinct site for viroporin inhibitors on the E protein of SARS-CoV-2. Virol J. 2023;20(1):142. DOI: 10.1186/s12985-023-02095-y
113. Saldivar-Espinoza B. et al. The mutational landscape of SARS-CoV-2. Int J Mol Sci. 2023;24(10):9072. DOI: 10.3390/ijms24109072
114. Tang Z. et al. Clinical characteristics and host immunity responses of SARS-CoV-2 Omicron variant BA.2 with deletion of ORF7a, ORF7b and ORF8. Virol J. 2023;20(1):106. DOI: 10.1186/s12985-023-02066-3
115. Miller A.N. et al. The SARS-CoV-2 accessory protein Orf3a is not an ion channel, but does interact with trafficking proteins. Elife. 2023;12:e84477. DOI: 10.7554/elife.84477
116. Zhang J. et al. SARS-CoV-2 ORF3a protein as a therapeutic target against COVID-19 and Long-term post-infection effects. Pathogens. 2024;13(1):75. DOI: 10.3390/pathogens13010075
117. Abbasian M.H. et al. Global landscape of SARS-CoV-2 mutations and conserved regions. J Transl Med. 2023; 21(1):152. DOI: 10.1186/s12967-023-03996-w
118. Papa G. et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021;17(1):e1009246. DOI: 10.1371/journal.ppat.1009246
119. Vela J.M. Repurposing sigma-1 receptor ligands for COVID-19 therapy? Front Pharmacol. 2020; 11:582310. DOI: 10.3389/fphar.2020.582310
120. Abatematteo F.S. et al. A conformational rearrangement of the SARS-CoV-2 host protein sigma-1 is required for antiviral activity: insights from a combined in-silico/in-vitro approach. Sci Rep. 2023; 13(1):12798. DOI: 10.1038/s41598-023-39662-w
121. Tasneem A. et al. Identification of potential therapeutic targets for COVID-19 through a structural-based similarity approach between SARS-CoV-2 and its human host proteins. Front Genet. 2024;15:1292280. DOI: 10.3389/fgene.2024.1292280
122. von Delft A. et al. Accelerating antiviral drug discovery: lessons from COVID-19. Nat Rev Drug Discov. 2023: 22(7):585-603. DOI: 10.1038/s41573-023-00692-8
На первых этапах поиск противовирусных соединений, блокирующих развитие вируса SARS-CoV-2, был направлен в основном на разрешенные к медицинскому применению лекарственные препараты (репозиционирование лекарств). Основное внимание уделялось главной протеазе вируса 3CLpro. Аминокислотная последовательность 3CLpro SARS-Cov-2 на 96% идентична последовательности этого фермента у SARS-CoV. 3CLpro представляет собой трехдоменную цистеиновую протеазу с каталитической диадой Cys145-His41, расположенной в щели между доменами I (остатки с 10 по 99) и II (остатки с сотого по 182). Домены I и II состоят из β-листов и образуют основную каталитическую субъединицу, тогда как домен III представляет собой компактный α-спиральный домен, соединенный с доменом II длинной линкерной петлей. Для активации фермента необходима гомодимеризация протомеров. Домен III (остатки со сто девяносто восьмого по 303) регулирует димеризацию 3CLpro посредством взаимодействия солевого мостика Glu-Arg. N-конец каждого протомера взаимодействует с Glu166 аналога и активирует карман S1 обоих протомеров для связывания субстрата. Моделирование молекулярной динамики 3CLpro показало, что мономерная форма, связанная с субстратом, делает структуру более жесткой и предотвращает вращение домена III, облегчая димеризацию фермента, необходимую для каталитической активности. Сравнение димера в связанном и несвязанном состояниях выявило наличие гидрофобного кластера, который аллостерически контролирует вращение домена III.
Поскольку главные протеазы 3CLpro вирусов SARS-CoV и SARS-CoV-2 очень схожи, разработанные ранее ингибиторы протеазы вируса SARS-CoV оказались активны и в отношении фермента нового коронавируса. Гомологов 3CLpro у человека нет. Учитывая важную роль фермента в процессинге полипротеинов и репликации вирусной РНК, он является наиболее изучаемой мишенью SARS-CoV-2. При этом в качестве места связывания для ингибирования рассматриваются как активный центр фермента, так и область контакта субъединиц.
Пространственная структура главной протеазы SARS-CoV-2 в комплексах с пептидомиметиками была расшифрована достаточно быстро (на 11 января 2021 года в PDB уже была доступна 421 структура 3CLpro SARS-CoV-2; в настоящее время число расшифрованных структур достигло 835).
Молекулярное моделирование применяется для исследования поведения и подвижности 3CLpro и для поиска новых ингибиторов этого белка. Детальный анализ структуры 3CLpro и изменения ее подвижности при различных условиях проводится методом молекулярной динамики [1]. Были исследованы подвижность активного центра фермента, появление в структуре скрытых аллостерических участков [2], влияние димеризации на структуру активного центра и др.
Большинство работ было проведено с использованием метода молекулярного докинга для поиска ингибиторов этого фермента. При этом применялись различные программы докинга, включая AutoDock, AutoDock Vina, DOCK, Glide и др. Сопоставление результатов докинга ингибиторов в 3CLpro с использованием различных программных продуктов показало, что наиболее точные результаты для нековалентных ингибиторов получаются с использованием программы Glide (Schrödinger), тогда как для ковалентных ингибиторов лучшей была программа EnzyDock [3]. Для нековалентных ингибиторов точность (воспроизведение лучшей по оценочной функции позы с RMSD < 2 Å от положения лиганда в кристалле) составила 26%, для ковалентных – 45%. Более точные результаты были получены для объемных, занимающих более одного субсайта, ингибиторов протеазы. Однако, было отмечено, что результаты докинга для 3CLpro надо интерпретировать с осторожностью, и для повышения корректности оценки необходимо проводить дополнительные исследования с использованием более точных методов предсказания аффинности, чем оценочные функции. В ряде работ по поиску новых ингибиторов с использованием методов докинга после отбора потенциальных лигандов по результатам докинга проводили исследования отобранных молекул с применением молекулярной динамики для оценки стабильности полученных комплексов и оценки аффинности методом MM-G(P)BSA [4-6].
В числе репозиционируемых лекарств, по результатам молекулярного моделирования рассматриваемых в качестве потенциальных ингибиторов 3CLpro, были антивирусные (паритапревир, саквинавир, ралтегравир и др.), противогрибковые (итраконазол), антибактериальные препараты (новобиоцин) [7] и даже витамины [8]. Наиболее известным ингибитором 3CLpro нового коронавируса является нирматрелвир (PF-07321332), разрешенный к применению в клинике в комбинации с ритонавиром (Paxlovid). По структуре – это пептидомиметик с четырьмя неканоническими аминокислотами, который является ковалентным ингибитором, связывающимся с каталитическим цистеином-145 главной протеазы 3CLpro. Недавно была опубликована обобщающая статья The COVID Moonshot Consortium, в рамках которого было сконструировано более 18 тысяч соединений. Свыше 2400 соединений было синтезировано, получено около пятисот пространственных структур комплексов основной протеазы SARS-CoV-2 с малыми молекулами и было проведено более десяти тысяч измерений ингибиторной активности [9], результаты которых оперативно выставлялись в свободный доступ. Эти исследования основаны на использовании подхода, получившего название Fragment-Based Drug Design (конструирование лекарств на основе фрагментов). Было изучено связывание фрагментов лекарственно-подобных соединений с главной протеазой SARS-CoV-2. При этом найдены фрагменты, связывающиеся с протеазой как за счет ковалентных, так и за счет нековалентных взаимодействий. При этом часть исследованных фрагментов взаимодействовали с 3CLpro вне активного центра. Информация по пространственным структурам комплексов, полученная в рамках выполнения этого проекта, была использована при разработке лекарственного препарата энситрелвир. Информация об экспериментально установленных количественных характеристиках активности ингибиторов 3CLpro и результатах клинических испытаний приведена на странице Репозиционирование.
В настоящее время большинство созданных ингибиторов относятся к пептидомиметикам, которые связываются с протеазой 3CLpro за счет ковалентных взаимодействий. Однако такие соединения обладают рядом недостатков, включая плохую проницаемость через клеточную мембрану, высокую метаболическую стабильность, возможные побочные эффекты за счет реакционной группы, нацеленной на каталитический цистеин. Поэтому в последнее время повысился интерес к разработке нековалентных ингибиторов главной протеазы коронавируса SARS-CoV-2 [10-12].
В дальнейшем, с расшифровкой пространственных структур других белков вируса, активно начались работы и в этих направлениях. Ниже представлен обзор сведений по молекулярному моделированию для других мишеней SARS-CoV-2.
Основными методами, основанными на структуре мишени, которые используются в процессе поиска лекарств с целью терапии COVID-19, являются молекулярный докинг и моделирование молекулярной динамики. Для предварительного отбора наиболее перспективных соединений иногда применяют методы глубокого машинного обучения и модели фармакофоров.
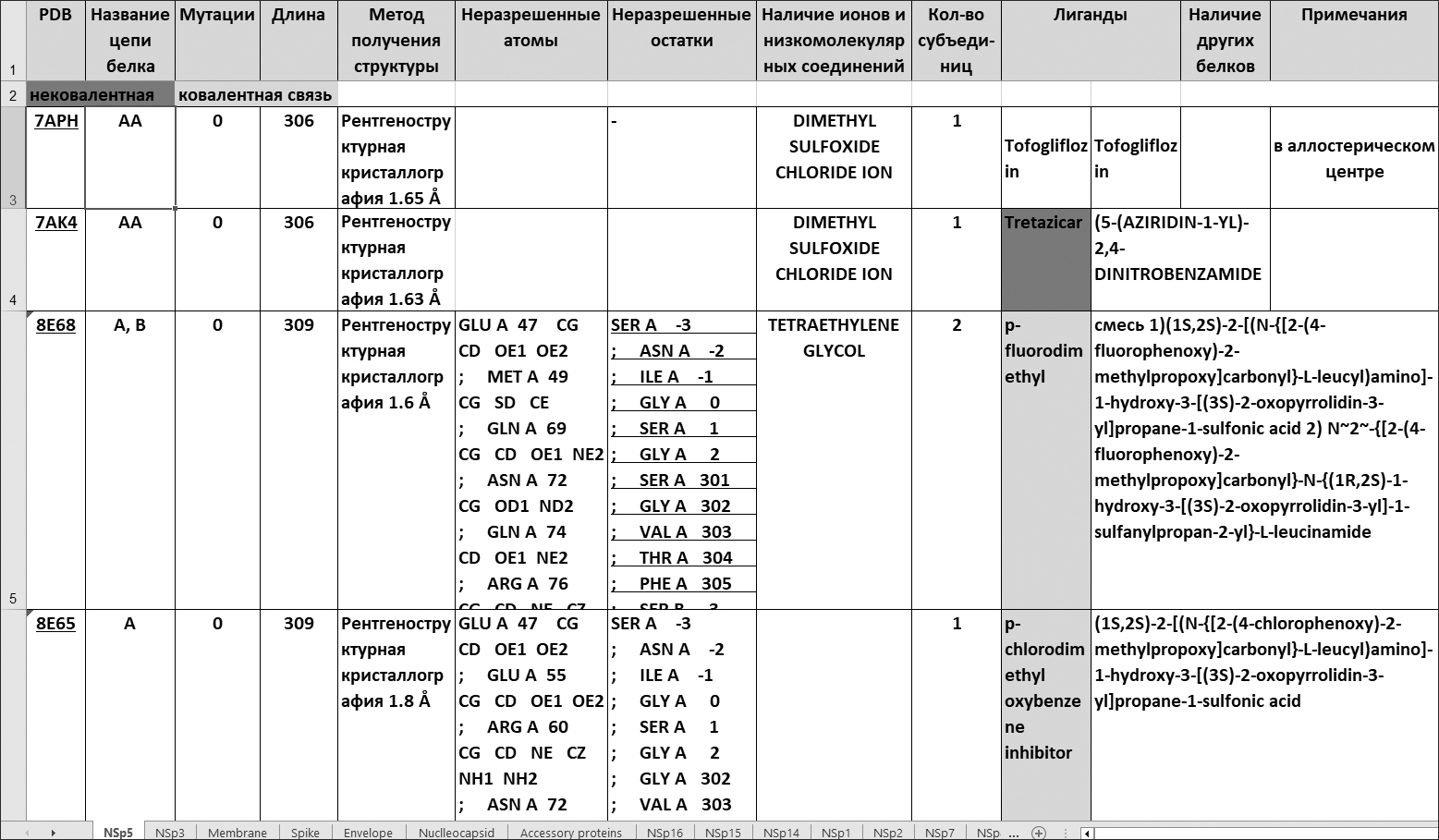
На основе анализа БД NCBI и PDB мы подготовили сводный перечень пространственных структур белков – молекулярных мишеней, ингибирование которых может быть использовано для терапии инфекции SARS-CoV-2/COVID-19. Наличие 3D структуры белка, желательно в комплексе с лигандом, является необходимым условием для применения методов дизайна лекарств на основе структуры мишени.
Для применения компьютерных методов, основанных на структуре мишени, мы провели отбор релевантных структур на основе следующих критериев:
1) Разрешение полученной структуры должно быть не хуже 3 Å – структуры с более низким разрешением не были включены в таблицу.
2) Исключались структуры, содержащие лишь небольшие фрагменты интересующих белков, структуры белков с большим количеством (более 30%) неразрешенных аминокислотных остатков.
Количество отобранных нами для включения в перечень пространственных структур белков и их число для соответствующего белка в БД PDB представлены в Таблице 1.
| Белок SARS-CoV-2 | Отобрано | Всего в PDB |
|---|---|---|
| Nsp1 | 14 | 28 |
| Nsp2 | 1 | 3 |
| Nsp3 | 526 | 526 |
| Nsp5 | 729 | 835 |
| Nsp7 | 8 | 50 |
| Nsp8 | 4 | 56 |
| Nsp9 | 8 | 13 |
| Nsp10 | 6 | 45 |
| Nsp12 | 20 | 45 |
| Nsp13 | 55 | 72 |
| Nsp14 | 75 | 70 |
| Nsp15 | 46 | 50 |
| Nsp16 | 39 | 32 |
| Spike | 142 | 850 |
| Envelope | 3 | 8 |
| Membrane | 2 | 3 |
| Nucleocapsid | 39 | 46 |
| Accessory proteins | 10 | 10 |
| ВСЕГО | 1722 | 2639 |
Как видно из таблицы, для большей части белков, число отобранных для включения в перечень пространственных структур белков меньше их числа в БД PDB. Это объясняется тем, что отбор производился с учетом фильтрации согласно приведенным выше критериям. Однако, для белков Nsp14 и Nsp16 сложилась обратная ситуация – количество структур в таблице превышает таковое значение в БД PDB. Это объясняется тем, что структуры комплексов этих белков с Nsp10 были записаны в структуры Nsp10 БД PDB, однако, кажется целесообразным перенести их в разделы белков Nsp14 и Nsp16 нашей таблицы, поскольку функции этих белков являются ключевыми для синтеза вирусной РНК, а Nsp10 выступает в роли активатора этих функций.
Отличительные особенности представления сведений в нашем перечне по сравнению с исходными форматами представления информации в БД NCBI и PDB следующие:
1) информация в наших таблицах представлена в компактной, лаконичной форме, но достаточна по объему для принятия решения о том, отвечает ли данная структура целям исследователя;
2) в таблицах особо отмечены лиганды, которые соединены с белком ковалентными связями;
3) в явном виде представлена информация о мутациях, имеющихся в структуре, неразрешенных атомах и остатках.
Такого рода таблицы сформированы нами для каждого из белков SARS-CoV-2, для которых известны пространственные структуры. Блоки информации, представленные в таблицах, как правило, совпадают для всех белков, например, код PDB, длина цепи, мутации, и т.д.
Фрагмент сводной таблицы по отобранным нами из PDB структурам белков-мишеней SARS-CoV-2 представлен в Таблице 2.
Полностью таблицы по доступным для молекулярного моделирования пространственным структурам SARS-CoV-2 представлены в разделе Библиотека.
Мы провели анализ наиболее широко используемых подходов и результатов молекулярного моделирования, направленного на поиск потенциальных антикоронавирусных соединений, в работах 2020-2023 гг.
Структурные формулы активных фармацевтических субстанций обычно получают из DrugBank, ZINC, PubChem, GenBank, MTiOpenScreen; либо источник в публикации не указан. Нередко для виртуального скрининга используют библиотеки, содержащие информацию обо всех лекарственных препаратах. Однако, в последнее время более широко стали применять сфокусированные библиотеки, например, соединения с антибактериальной или антивирусной активностью [13], известные ингибиторы для определенного типа ферментов [14-16].
Необходимо отметить, что в большинстве последних публикаций моделирование методами докинга дополняется результатами молекулярной динамики с оценкой активности ингибиторов в отношении белков коронавируса (см. например, [17]).
Наиболее популярными мишенями являются S-белок, РНК-зависимая РНК полимераза (RdRp), главная (3CLpro) и папаиноподобная (PLpro) протеазы. Но встречаются также работы, в которых в качестве мишеней рассматривают белки E [18, 19], N [20], S [21-23], Nsp3 [24, 25], Nsp4 [26, 27], Nsp7 [28], Nsp9 [29], Nsp13 [30, 31], Nsp15 [24] и Nsp16 [30-32].
Наиболее часто используемыми для докинга компьютерными программами являются: AutoDock [33], Autodock Vina [34], Glide [35], MOE [36], Dock [37], ICM [38].
В ряде работ применяют методы глубокого машинного обучения (Deep Machine Learning) с целью предварительной селекции наиболее перспективных фармацевтических субстанций для молекулярного моделирования и поиска новых потенциально активных соединений [19, 39-44].
Для предварительного отбора соединений-лидеров применяют программы докинга, а оценку стабильности комплексов соединений с белком проводят при помощи молекулярной динамики; расчетные результаты проверяют путем оценки ингибирующей активности отобранных фармацевтических субстанций в отношении мишени in vitro [45].
Моделирование молекулярной динамики чаще всего проводят с помощью Gromacs [46], но используют также и Desmond [47], Amber [48], NAMD [49]. Для построения пространственной структуры белка применяют SWISS-MODEL [50] или Modeller [51]. Во многих работах для оценки энергии связывания в системе белок-лиганд применяется метод MM-P(G)BSA [52].
Несмотря на то, что поиск антикоронавирусных соединений in silico и в экспериментах in vitro для подавляющего большинства разрешенных к применению лекарств уже был проведен неоднократно, новые работы такого рода публикуются до сих пор. В последних исследованиях виртуальный скрининг намного чаще сопровождается экспериментальной проверкой компьютерных предсказаний. В результате таких работ были найдены дополнительные соединения, ингибирующие белки SARS-CoV-2, но их активность обычно невысока [21, 26, 41, 53-55], что требует последующей оптимизации структуры и свойств выявленных «хитов».
В последнее время интересным направлением исследований стало конструирование искусственных белков, которые должны связываться с поверхностными белками вирусных частиц, и таким образом нейтрализовать вирус [56-58].
Для быстрой проверки гипотез по репозиционированию представляется необходимым создание коллекции физических образцов лекарств и соединений, прошедших первую фазу клинических испытаний. Количество каждого соединения должно быть достаточно для проведения полного цикла проверки на эффективность в системах in vitro и in vivo.
Необходимо отметить также активные работы по детальному изучению механизмов взаимодействия антикоронавирусных препаратов (фавипиравир, молнупиравир, ремдесивир, нирматрелвир) и их активных метаболитов с белками вируса [59-63]. Основное направление этих исследований – оптимизация структуры и свойств этих соединений на основе детального знания механизмов их взаимодействия с мишенями с целью создания более эффективных препаратов.
Появление новых штаммов вируса заставило обратить пристальное внимание на обусловленные мутациями в белках-мишенях изменения (1) структуры самих белков-мишеней, (2) взаимодействий белков вируса с белками человека [64-66], и (3) аффинности лекарственных препаратов к вирусным белкам [21, 67-69].
Уже на первых этапах работ по поиску лекарственных препаратов, блокирующих рост и размножение вируса SARS-CoV-2, белки человека, которые задействованы в цикле развития вируса, рассматривались как потенциальные мишени. Первыми такими белками были ACE2 и протеаза TMPRSS2 [70-72]. К настоящему моменту к ним добавился еще ряд потенциальных мишеней (басигин, фурин; нейропилин-1) [73-75]. Повышенный интерес к таким мишеням во многом объясняется высокой мутационной способностью SARS-CoV-2, поэтому соединения, действующие на белки хозяина, рассматриваются как перспективный вариант снизить влияние этой изменчивости вируса. В то же время нельзя исключить, что воздействие на вовлеченные в жизненный цикл SARS-CoV-2 белки человека может приводить к возникновению побочных эффектов.
Критический обзор использования методов молекулярного моделирования в 2020-2022 гг. приведен в уже упомянутой в разделе 1 публикации [76]. В этой работе также приведены рекомендации по валидации применяемых расчетных методов, что позволит повысить качество получаемых результатов.
Таким образом, анализ литературы показывает, что в настоящее время исследования, направленные на изучение вируса SARS-CoV-2, продолжают активно развиваться. Одновременно с традиционными направлениями по поиску и конструированию новых низкомолекулярных соединений, взаимодействующих с белками вируса, все большее развитие получают исследования, отражающие новые вызовы в связи с мутациями и адаптацией вируса к новым условиям. Появляются новые направления, такие как конструирование белков и аптамеров к белкам-мишеням, а также повышается интерес к разработке соединений, взаимодействующих с белками человека. Основой для последнего направления является надежда, что к таким соединениям у вируса не будет возможности адаптироваться.
Цитируемая литература:
1. Padhi A.K. et al. Accelerating COVID-19 research using molecular dynamics simulation. J. Phys. Chem. B. 2021;125(32):9078-9091. DOI: 10.1021/acs.jpcb.1c04556
2. Sztain T. et al. Elucidation of cryptic and allosteric pockets within the SARS-CoV-2 main protease. J. Chem. Inf. Model. 2021;61(7):3495-3501. DOI: 10.1021/acs.jcim.1c00140
3. Zev S. et al. Benchmarking the ability of common docking programs to correctly reproduce and score binding modes in SARS-CoV-2 protease Mpro. J. Chem. Inf. Model., 2021;61(6):2957-2966. DOI: 10.1021/acs.jcim.1c00263
4. Rubio-Martínez J. et al. Discovery of diverse natural products as inhibitors of SARS-CoV-2 Mpro protease through virtual screening. J Chem. Inf. Model. 2021;61(12):6094-6106. DOI: 10.1021/acs.jcim.1c00951
5. Gupta A., Zhou H.X. Profiling SARS-CoV-2 main protease (MPRO) binding to re-purposed drugs using molecular dynamics simulations in classical and neural network-trained force fields. ACS Comb Sci. 2020;22(12):826-832. DOI: 10.1021/acscombsci.0c00140
6. Marimuthu P. et al. Mechanistic insights into SARS-CoV-2 main protease inhibition reveals hotspot residues. J Chem Inf Model. 2021;61(12):6053-6065. DOI: 10.1021/acs.jcim.1c00928
7. Wu C. et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020;10(5): 766-788. DOI: 10.1016/j.apsb.2020.02.008
8. Kandeel M., Al-Nazawi M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020;251:117627. DOI: 10.1016/j.lfs.2020.117627
9. Boby M.L. et al. Open science discovery of potent noncovalent SARS-CoV-2 main protease inhibitors. Science. 2023;382(6671):eabo7201. DOI: 10.1126/science.abo7201
10. Unoh Y. et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J Med Chem. 2022;65(9):6499-6512. DOI: 10.1021/acs.jmedchem.2c00117
11. Gao S. et al. Discovery and crystallographic studies of trisubstituted piperazine derivatives as non-covalent SARS-CoV-2 main protease inhibitors with high target specificity and low toxicity. J Med Chem. 2022;65(19):13343-13364. DOI: 10.1021/acs.jmedchem.2c01146
12. Gao S. et al. Design, synthesis, and biological evaluation of trisubstituted piperazine derivatives as noncovalent severe acute respiratory syndrome coronavirus 2 main protease inhibitors with improved antiviral activity and favorable druggability. J Med Chem. 2023;66(23):16426–16440. DOI: 10.1021/acs.jmedchem.3c01876
13. Tyagi R. et al. Drug repurposing approach to identify therapeutics by screening pathogen box exploiting SARS-CoV-2 main protease. Chem Biodivers. 2023;20(2):e202200600. DOI: 10.1002/cbdv.202200600
14. Rampogu S. et al. Repurposing and computational design of PARP inhibitors as SARS-CoV-2 inhibitors. Sci Rep. 2023;13(1):10583. DOI: 10.1038/s41598-023-36342-7
15. Boytz R. et al. Anti-SARS-CoV-2 activity of targeted kinase inhibitors: Repurposing clinically available drugs for COVID-19 therapy. J Med Virol. 2023;95(1):e28157. DOI: 10.1002/jmv.28157
16. El-Hddad S., Sobhy M., Ayoub A. et al. In silico molecular docking, dynamics simulation and repurposing of some VEGFR-2 inhibitors based on the SARS-CoV-2-main-protease inhibitor N3. J Biomol Struct Dyn. 2023;41(19):9267-9281. DOI: 10.1080/07391102.2022.2148000
17. Freidel M.R., Armen R.S. Modeling the structure–activity relationship of arbidol derivatives and other SARS-CoV-2 fusion inhibitors targeting the S2 segment of the spike protein. J Chem Inform Model. 2021;61(12):5906-5922. DOI: 10.1021/acs.jcim.1c01061
18. Dey D. et al. In silico identification of Tretinoin as a SARS-CoV-2 envelope (E) protein ion channel inhibitor. Comput Biol Med. 2020;127:104063. DOI: 10.1016/j.compbiomed.2020.104063
19. Das G. et al. Repurposed drugs and nutraceuticals targeting envelope protein: A possible therapeutic strategy against COVID-19. Genomics. 2021;113(1 Pt 2):1129-1140. DOI: 10.1016/j.ygeno.2020.11.009
20. Kim J. et al. Abiraterone acetate attenuates SARS-CoV-2 replication by interfering with the structural nucleocapsid protein. Biomol Ther. (Seoul). 2022;30(5)427-434. DOI: 10.4062/biomolther.2022.037
21. Fidan O. et al. Discovery of adapalene and dihydrotachysterol as antiviral agents for the Omicron variant of SARS-CoV-2 through computational drug repurposing. Mol Divers. 2023;27(1):463-475. DOI: 10.1007/s11030-022-10440-6
22. Sawang N. et al. Biophysical interpretation of evolutionary consequences on the SARS-CoV-2 main protease through molecular dynamics simulations and network topology analysis. J Phys Chem B. 2023;127(11):2331-2343. DOI: 10.1021/acs.jpcb.2c08312
23. Wang Q. et al. In silico discovery of small molecule modulators targeting the Achilles' heel of SARS-CoV-2 spike protein. ACS Cent Sci. 2023;9(2):252-265. DOI: 10.1021/acscentsci.2c01190
24. Kumar S. et al. Multi-targeting approach for nsp3, nsp9, nsp12 and nsp15 proteins of SARS-CoV-2 by Diosmin as illustrated by molecular docking and molecular dynamics simulation methodologies. Methods. 2021;195:44-56. DOI: 10.1016/j.ymeth.2021.02.017
25. Singh A.K. et al. Identification of FDA approved drugs and nucleoside analogues as potential SARS-CoV-2 A1pp domain inhibitor: An in silico study. Comput Biol Med. 2021;130:104185. DOI: 10.1016/j.compbiomed.2020.104185
26. Chakraborty J, Maity A, Sarkar H. A systematic drug repurposing approach to identify promising inhibitors from FDA-approved drugs against nsp4 protein of SARS-CoV-2. J Biomol Struct Dyn. 2023;41(2):550-559. DOI: 10.1080/07391102.2021.2009033
27. Azizogli A.R. et al. Scalable inhibitors of the Nsp3-Nsp4 coupling in SARS-CoV-2. ACS Omega. 2023;8(6):5349-5360. DOI: 10.1021/acsomega.2c06384
28. Gu Y. et al. Drug-repurposing screening identifies a gallic acid binding site on SARS-CoV-2 non-structural protein 7. ACS Pharmacol Transl Sci. 2023;6(4):578-586. DOI: 10.1021/acsptsci.2c00225
29. Mehyar N. et al. Discovery of Zafirlukast as a novel SARS-CoV-2 helicase inhibitor using in silico modelling and a FRET-based assay. SAR QSAR Environ Res. 2021;32(12):963–983. DOI: 10.1080/1062936x.2021.1993995
30. Vijayan V. et al. Identification of promising drug candidates against NSP16 of SARS-CoV-2 through computational drug repurposing study. J Biomol Struct Dyn. 2021;39(17):6713-6727. DOI: 10.1080/07391102.2020.1802349
31. Samdani M.N. et al. Targeting SARS-CoV-2 non-structural protein 13 via helicase-inhibitor-repurposing and non-structural protein 16 through pharmacophore-based screening. Mol Divers. 2023;27(3):1067-1085. DOI: 10.1007/s11030-022-10468-8
32. Elmorsy M.A. et al. In silico screening of potent inhibitors against COVID-19 key targets from a library of FDA-approved drugs. Environ Sci Pollut Res Int. 2022;29(8):12336-12346. DOI: 10.1007/s11356-021-16427-4
33. Morris G.M. et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem. 2009;16: 2785-2791. DOI: 10.1002/jcc.21256
34. Trott O., Olson A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455-461. DOI: 10.1002/jcc.21334
35. Friesner R.A. et al. Extra precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49: 6177-6196. DOI: 10.1021/jm051256o
36. Vilar S. et al. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr Top Med Chem. 2008;8:1555-1572. DOI: 10.2174/156802608786786624
37. Oshiro C.M., Kuntz I.D., Dixon J.S. Flexible ligand docking using a genetic algorithm. J Comput-Aided Mol Design. 1995;9:113-130. DOI: 10.1007/bf00124402
38. Neves M.A. et al. Docking and scoring with ICM: the benchmarking results and strategies for improvement. J Comput-Aided Mol Des. 2012;26(6): 675-686. DOI: 10.1007/s10822-012-9547-0
39. Anwaar M.U. et al. Combined deep learning and molecular docking simulations approach identifies potentially effective FDA approved drugs for repurposing against SARS-CoV-2. Comput Biol Med. 2022;141:105049. DOI: 10.1016/j.compbiomed.2021.105049
40. Joshi T. et al. Predictive modeling by deep learning, virtual screening and molecular dynamics study of natural compounds against SARS-CoV-2 main protease. J Biomol Struct Dyn. 2021;39:6728-6746. DOI: 10.1080/07391102.2020.1802341
41. Yao Y. et al. A deep learning-based drug repurposing screening and validation for anti-SARS-CoV-2 compounds by targeting the cell entry mechanism. Biochem Biophys Res Commun. 2023;675:113-121. DOI: 10.1016/j.bbrc.2023.07.018
42. Cohen S. et al. Improved and optimized drug repurposing for the SARS-CoV-2 pandemic. PLoS One. 2023;18(3):e0266572. DOI: 10.1371/journal.pone.0266572
43. Joshi R.P. et al. AI-accelerated design of targeted covalent inhibitors for SARS-CoV-2. J Chem Inf Model. 2023;63(5):1438-1453. DOI: 10.1021/acs.jcim.2c01377
44. Saramago L.C. et al. AI-Driven discovery of SARS-CoV-2 main protease fragment-like inhibitors with antiviral activity in vitro. J Chem Inf Model. 2023;63(9):2866-2880. DOI: 10.1021/acs.jpcb.2c02365
45. Jang W.D. et al. Drugs repurposed for COVID-19 by virtual screening of 6,218 drugs and cell-based assay. Proc Natl Acad Sci USA. 2021;118(30):e2024302118. DOI: 10.1073/pnas.2024302118
46. Abraham M.J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19-25. DOI: 10.1016/j.softx.2015.06.001
47. Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2021. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2021. Ссылка
48. Case D.A. et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668-1688. DOI: 10.1002/jcc.20290
49. Phillips J.C. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys. 2020;153(4):044130. DOI: 10.1063/5.0014475
50. Waterhouse A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296-W303. DOI: 10.1093/nar/gky427
51. Fiser A., Sali A. Modeller: generation and refinement of homology-based protein structure models. Macromolecular Crystallography, Part D. Methods in Enzymology. 2003;374:461–491. DOI: 10.1016/s0076-6879(03)74020-8
52. Huo S. et al. Computational alanine scanning of the 1:1 human growth hormone-receptor complex. J Comput Chem. 2002;23(1):15-27. DOI: 10.1002/jcc.1153
53. Ahmed M.S. et al. FDA approved drugs with antiviral activity against SARS-CoV-2: From structure-based repurposing to host-specific mechanisms. Biomed Pharmacother. 2023;162:114614. DOI: 10.1016/j.biopha.2023.114614
54. Khan A.M. et al. Repurposing of US-FDA approved drugs against SARS-CoV-2 main protease (Mpro) by using STD-NMR spectroscopy, in silico studies and antiviral assays. Int J Biol Macromol. 2023;234:123540. DOI: 10.1016/j.ijbiomac.2023.123540
55. Ambrosio F.A. et al. Targeting SARS-CoV-2 main protease: A successful story guided by an in silico drug repurposing approach. J Chem Inf Model. 2023;63(11):3601-3613. DOI: 10.1021/acs.jcim.3c00282
56. Kalita P. et al. Computational protein design for COVID-19 research and emerging therapeutics. ACS Cent Sci. 2023;9(4):602-613. DOI: 10.1021/acscentsci.2c01513
57. Padhi A.K. et al. From de novo design to redesign: Harnessing computational protein design for understanding SARS-CoV-2 molecular mechanisms and developing therapeutics. J Phys Chem B. 2023;127(41):8717-8735. DOI: 10.1021/acs.jpcb.3c04542
58. Joshi A. et al. Computational approaches for development of engineered therapeutics against SARS-CoV-2. Biochemistry. 2023;62(3):669-671. DOI: 10.1021/acs.jpcb.2c02365
59. Samanta P.N. et al. Elucidating atomistic insight into the dynamical responses of the SARS-CoV-2 main protease for the binding of remdesivir analogues: Leveraging molecular mechanics to decode the inhibition mechanism. J Chem Inf Model. 2023;63(11):3404-3422. DOI: 10.1021/acs.jcim.3c00105
60. Bhati A.P. et al. Long time scale ensemble methods in molecular dynamics: ligand-protein interactions and allostery in SARS-CoV-2 targets. J Chem Theory Comput. 2023;19(11):3359-3378. DOI: 10.1021/acs.jctc.3c00020
61. Chan H.T.H. et al. Dynamical nonequilibrium molecular dynamics simulations identify allosteric sites and positions associated with drug resistance in the SARS-CoV-2 Main Protease. JACS Au. 2023;3(6):1767-1774. DOI: 10.1021/jacsau.3c00185
62. You W., Chen Y. Exploring the inhibition mechanism of SARS-CoV-2 main protease by ebselen: A molecular docking, molecular dynamics simulation and DFT approach. J Comput Chem. 2023;44(26):2086-2095. DOI: 10.1002/jcc.27181
63. Xu L., Xie L., Zhang D. et al. Elucidation of Binding Features and Dissociation Pathways of Inhibitors and Modulators in SARS-CoV-2 Main Protease by Multiple Molecular Dynamics Simulations. Molecules. 2022;27(20):6823. DOI: 10.3390/molecules27206823
64. Bhadane R., Salo-Ahen O.M.H. High-Throughput Molecular Dynamics-Based Alchemical Free Energy Calculations for Predicting the Binding Free Energy Change Associated with the Selected Omicron Mutations in the Spike Receptor-Binding Domain of SARS-CoV-2. Biomedicines. 2022;10(11):2779. DOI: 10.3390/biomedicines10112779
65. Diessner E.M. et al. Mutation effects on structure and dynamics: Adaptive evolution of the SARS-CoV-2 main protease. Biochemistry. 2023;62(3):747-758. DOI: 10.1021/acs.biochem.2c00479
66. Vergara N.G. et al. Entropic overcompensation of the N501Y mutation on SARS-CoV-2 S binding to ACE2. J Chem Inf Model. 2023;63(2):633-642. DOI: 10.1021/acs.jcim.2c01246
67. Clayton J. et al. Integrative approach to dissect the drug resistance mechanism of the H172Y mutation of SARS-CoV-2 main protease. J Chem Inf Model. 2023;63(11):3521-3533. DOI: 10.1021/acs.jcim.3c00344
68. Mohamed E.A.R. et al. In silico prediction of potential inhibitors for SARS-CoV-2 Omicron variant using molecular docking and dynamics simulation-based drug repurposing. J Mol Model. 2023;29(3):70. DOI: 10.1007/s00894-023-05457-z
69. Jiang H. et al. Evaluation of the inhibition potency of nirmatrelvir against main protease mutants of SARS-CoV-2 variants. Biochemistry. 2023;62(13):2055-2064. DOI: 10.1021/acs.biochem.3c00075
70. Yang R. et al. Identification of potential TMPRSS2 inhibitors for COVID-19 treatment in Chinese medicine by computational approaches and surface plasmon resonance technology. J Chem Inf Model. 2023;63(10):3005-3017. DOI: 10.1021/acs.jcim.2c01643
71. Peiffer A.L. et al. TMPRSS2 inhibitor discovery facilitated through an in silico and biochemical screening platform. ACS Med Chem Lett. 2023;14(6):860-866. DOI: 10.1021/acsmedchemlett.3c00035
72. Mantzourani C., Vasilakaki S., Gerogianni V.E. et al. The discovery and development of transmembrane serine protease 2 (TMPRSS2) inhibitors as candidate drugs for the treatment of COVID-19. Expert Opin Drug Discov. 2022;17(3):231-246. DOI: 10.1080/17460441.2022.2029843
73. Yang Z. et al. Repurposing niclosamide as a novel anti-SARS-CoV-2 drug by restricting entry protein CD147. Biomedicines. 2023;11(7):2019. DOI: 10.3390/biomedicines11072019
74. Devi K.P. et al. A perspective on the applications of furin inhibitors for the treatment of SARS-CoV-2. Pharmacol Rep. 2022;74(2):425-430. DOI: 10.1007/s43440-021-00344-x
75. Afiadenyo M. et al. Computational screening of neuropilin-1 unveils novel potential anti-SARS-CoV-2 therapeutics. Chem Biodivers. 2023:e202301227. DOI: 10.1002/cbdv.202301227
76. Gentile F. et al. Surely you are joking, Mr Docking! Chem Soc Rev. Chem Soc Rev. 2023;52(3):872-878. DOI: 10.1039/d2cs00948j